Врожденные заболевания обмена веществ | Сант Жоан де Деу
Что такое наследственные болезни обмена веществ (врожденные метаболические заболевания)
Патологии, также известные как врожденные нарушения метаболизма, — это заболевания, причина которых кроется в генетическом изменении белка или фермента, в результате чего блокируется определенный процесс метаболизма. Такая блокировка влияет на нормальное функционирование некоторых клеток и органов и проявляется рядом симптомов, различных у каждого пациента. Среди таких симптомов могут встречаться разные виды неврологических синдромов.
Эта группа патологий очень обширна, однако ее можно систематизировать с помощью действующей классификации, которая в данный момент претерпевает значительные изменения ввиду того, что сегодня мы располагаем гораздо большими знаниями о базовых механизмах развития таких патологий. Ниже приведены основные группы патологий, составленные на основании типа поражения организма при каждой из них.
Врожденное нарушение метаболизма малых молекул
Влияют на промежуточный метаболизм. Сюда входят аминоацидопатии (фенилкетонурия и пропионовая ацидурия). Также сюда входит нарушения обмена углеводов или нейромедиаторов и нейромодуляторов.
Врожденное нарушение энергетического обмена
Характеризуется недостаточной выработкой и использованием энергии. Сюда входят митохондриальные заболевания, недостаточная выработка пирувата или глюкозы (в мышцах или печени) и т. д.
Врожденное нарушение метаболизма сложных молекул
Группа заболеваний, которые препятствуют синтезу больших молекул. Они проявляются в виде постоянных симптомов, не связанных с питанием. Сюда входят лизосомные (мукополисахаридоз, олигосахаридоз, сфинголипидоз и т. д.), пероксисомальные (синдром Цельвегера, адренолейкодистрофия, сцепленная с хромосомой Х) заболевания и врожденные нарушения гликозилирования, а также другие врожденные нарушения метаболизма.
Метаболический синдром — нарушение обмена веществ
«Метаболический синдром»– проблема, которая набирает обороты во всем Мире. Подробнее о том, что такое метаболический синдром, как с ним бороться расскажет Красильникова Марина Олеговна, врач-гастроэнтеролог, гепатолог сети многопрофильных клиник ДИАЛАЙН, кандидат медицинский наук и куратор программы по ведению пациентов с метаболическим синдромом.
Подробнее о том, что такое метаболический синдром, как с ним бороться расскажет Красильникова Марина Олеговна, врач-гастроэнтеролог, гепатолог сети многопрофильных клиник ДИАЛАЙН, кандидат медицинский наук и куратор программы по ведению пациентов с метаболическим синдромом.
— Что такое метаболический синдром?
— Метаболический синдром— одна из наиболее актуальных проблем современной медицины, связанная с ведением нездорового образа жизни. Метаболический синдром — это комплекс нарушений обмена веществ, при котором повышен риск развития сердечно-сосудистых заболеваний и диабета 2 типа. Снижение физической активности и высококалорийный характер питания являются главными причинами того, что заболеваемость метаболическим синдромом растет. На данный момент им страдает около 25% населения.
— Как определить его наличие?
— Предпосылками для появления метаболического синдрома являются малоподвижный образ жизни, избыток массы тела и ожирение, артериальная гипертензия, наличие ишемическая болезнь сердца, диабет 2 типа и некоторые другие заболевания. Важно вовремя диагностировать данное заболевание и начать лечение. Возможны следующие жалобы при метаболическом синдроме — повышенная утомляемость, апатия, одышка, повышенный аппетит, жажда, учащенное мочеиспускание, головная боль, ночная потливость, вспышки ярости, исчезающие после приема пищи, слабость, сонливость после мясной пищи. Основной признак наличия метаболического синдрома центральный (абдоминальный) тип ожирения—т.е. окружность талии более 80 см. у женщин и более 94 см. у мужчин, а также индекс массы тела более 25. Дополнительные критерии- определяет специалист: артериальное давление больше 140/90 мм рт. ст., повышение уровня ТТГ(тиреотропный гормон), изменение липидного спектра, гипогликемия натощак.
Важно вовремя диагностировать данное заболевание и начать лечение. Возможны следующие жалобы при метаболическом синдроме — повышенная утомляемость, апатия, одышка, повышенный аппетит, жажда, учащенное мочеиспускание, головная боль, ночная потливость, вспышки ярости, исчезающие после приема пищи, слабость, сонливость после мясной пищи. Основной признак наличия метаболического синдрома центральный (абдоминальный) тип ожирения—т.е. окружность талии более 80 см. у женщин и более 94 см. у мужчин, а также индекс массы тела более 25. Дополнительные критерии- определяет специалист: артериальное давление больше 140/90 мм рт. ст., повышение уровня ТТГ(тиреотропный гормон), изменение липидного спектра, гипогликемия натощак.
— Многие люди живут и не подозревают, что причиной ухудшения их состояния может быть метаболический синдром. Расскажите подробнее о том, что могут предложить в клиниках по диагностике и профилактике метаболического синдрома?
— В наших клиниках разработаны чек-апы – специальные комплексные программы обследования, которые позволят в короткий срок диагностировать наличие метаболического синдрома, своевременно назначить лечение и подобрать индивидуальную программу питания. Остановлюсь на чек-апе «метаболический синдром расширенный + ТТГ». Что он в себя включает: анализ ТТГ(тиреотропный гормон)- это гормон, определяющий функцию щитовидной железы и обмен веществ. Так же проводится ЭКГ и биоимпедансометрия -определение процентного соотношения воды, мышечной и жировой ткани для выявления метаболического синдрома.
Остановлюсь на чек-апе «метаболический синдром расширенный + ТТГ». Что он в себя включает: анализ ТТГ(тиреотропный гормон)- это гормон, определяющий функцию щитовидной железы и обмен веществ. Так же проводится ЭКГ и биоимпедансометрия -определение процентного соотношения воды, мышечной и жировой ткани для выявления метаболического синдрома.
— Как это происходит на практике и в какой срок?
— Пациент проходит лабораторные и инструментальные обследования, после чего следует консультация специалиста с назначением лечения и подбором программы питания. Срок прохождения данного чек-апа два дня.
Если вы обнаружили у себя признаки метаболического синдрома, не стоит затягивать, а лучше своевременно обратиться к специалисту.
Редкие эндокринные и метаболические заболевания
Редкие (орфанные) эндокринные и метаболические заболевания — это группа болезней и синдромов, связанных с неправильным функционированием одной или нескольких желез внутренней секреции и нарушениями метаболизма.
Всего насчитывается около 500 таких заболеваний, тем не менее, они затрагивают сотни тысяч человек во всем мире.
Симптомы, причины возникновения и клиническая картина редких эндокринных и метаболических заболеваний и синдромов могут сильно отличаться. Их сложно профилактировать, диагностировать, оценивать динамику течения и лечить. В большинстве случаев лекарственной терапии, способной устранить причину развития редкого эндокринного или метаболического заболевания, вовсе не существует.
Объединяет эту группу патологий два условия: разрушительное воздействие на работу органов эндокринной системы и нарушения метаболизма.
- болезнь Аддисона (гипокортицизм, хроническая надпочечниковая недостаточность)
- гиперадренализм
- гиперпаратиреоз
- феохромоцитома
- гестационный несахарный диабет
- порфирия
- амилоидоз наследственный
- болезнь Паркинсона 9 типа
- Болезнь Вильсона — Коновалова (гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона)
- акромегалия
- АКТГ-секретирующая аденома гипофиза
- АКТГ-независимая макронодулярная гиперплазия надпочечников
- аутоиммунный полигландулярный синдром 1, 2, 3 типа
- синдром Карпентера
- синдром Кушинга
- гигантизм
- гранулематозный гипофизит
- дефицит гормона роста
- наследственная параганглиома-феохромоцитома
- гипопаратиреоз
- гипопитуитаризм
- синдром Каллмана
- пролактинома
- псевдогипопаратиреоз
- дисгенез щитовидной железы
- изолированный дефицит ТТГ
- периферическая резистентность к гормонам щитовидной железы
- множественная эндокринная неоплазия (типы 1, 2, 2А, 2В)
- MODY (диабет зрелого типа у молодых)
- болезнь Гиппеля — Линдау (цереброретинальный ангиоматоз, VHL)
- синдром Уотерхауса — Фридериксена
- амилоидоз (AL, ATTR, AA)
- семейные периодические лихорадки (периодическая болезнь, криопиринопатии, синдром Макла — Уэллса, гипериммуноглобулинемия D, TRAPS-синдром)
- олигосекреторные плазмоклеточные дискразии (TEMPI, POEMS, синдром Шницлера)
Преимущества лечения редких эндокринных и метаболических заболеваний в клинике Рассвет
Редкие эндокринные и метаболические заболевания и синдромы часто относят к «медицинским загадкам».
Лечение орфанных заболеваний требует не только профессионального практического опыта, но и высокой самоподготовки, постоянного изучения редких болезней.
Врачи клиники Рассвет обладают всеми необходимыми знаниями и навыками, помогающими диагностировать и лечить редкие эндокринные и метаболические заболевания, а также отличать их от типичных болезней с необычной картиной течения.
Отдел диагностики и лечения метаболических заболеваний
Метаболические заболевания – это группа заболеваний, связанных с нарушением обмена веществ (метаболизма). К метаболическим заболеваниям относятся сахарный диабет, ожирение, нарушение липидного обмена (дислипидемия) и другие.
Сахарный диабет (СД) – тяжелое хроническое заболевание, при несоблюдении определенных правил питания, лечения и самоконтроля, может привести к тяжелым осложнениям и инвалидности. Ожирение-хроническое заболевание, характеризующееся увеличением массы жировой ткани, нарушением обмена веществ, поражением органов и систем и развитием пищевой зависимости. По данным ВОЗ в 2016 году в мире насчитывалось около 2 миллиардов взрослых старше 18 лет, которые имели лишний вес: из них более 650 млн страдали ожирением. Ожирение ассоциируется с большим количеством заболеваний, в том числе с сахарным диабетом и развитием онкопатологии. Поражение нервной системы одно из наиболее распространенных и наиболее угрожающих осложнений, как сахарного диабета, так и ожирения.
Ожирение-хроническое заболевание, характеризующееся увеличением массы жировой ткани, нарушением обмена веществ, поражением органов и систем и развитием пищевой зависимости. По данным ВОЗ в 2016 году в мире насчитывалось около 2 миллиардов взрослых старше 18 лет, которые имели лишний вес: из них более 650 млн страдали ожирением. Ожирение ассоциируется с большим количеством заболеваний, в том числе с сахарным диабетом и развитием онкопатологии. Поражение нервной системы одно из наиболее распространенных и наиболее угрожающих осложнений, как сахарного диабета, так и ожирения.
Учитывая чрезвычайную актуальность проблем ожирения, сахарного диабета и его осложнений, основная деятельность сотрудников отдела диагностики и лечения метаболических заболеваний направлена на изучение состояния пациентов и поиски путей оптимизации их лечения. Сотрудники отдела проводят анализ суточного колебания гликемии с помощью современного оборудования (CGMS). Также в отделе проводится установка инсулиновых дозаторов (помпа Терапия) с последующим обучением и контролем пациентов.
Большинство сотрудников отдела имеют научную степень кандидата медицинских наук, постоянно проходят стажировку и обучение за рубежом, для совершенствования навыков, в дальнейшем применяют полученные знания в работе с пациентами.
Ожирение и метаболические нарушения у пациентов с COVID-19
Оригинал: Nature Reviews Endocrinology
Автор: Stefan et al.
Опубликовано: Nature Reviews Endocrinology 23.04.2020 (www.nature.com/nrendo)
Перевод: Елена Алексеенкова, Фонд профилактики рака
АннотацияПо предварительным данным, у людей с ожирением выше риск тяжелого течения COVID-19. Однако доступной на сегодняшний день информации о метаболических параметрах (ИМТ, концентрации глюкозы и инсулина в сыворотке крови) пациентов с COVID-19 недостаточно. Для лучшего понимания течения заболевания, совершенствования лечения и ухода за пациентами с COVID-19 необходимы дополнительные данные.
Введение
В Китае у пациентов, инфицированных SARS-CoV-2, возраст старше 65 лет и наличие сопутствующих заболеваний оказались ассоциированы с более тяжелым течением COVID-19.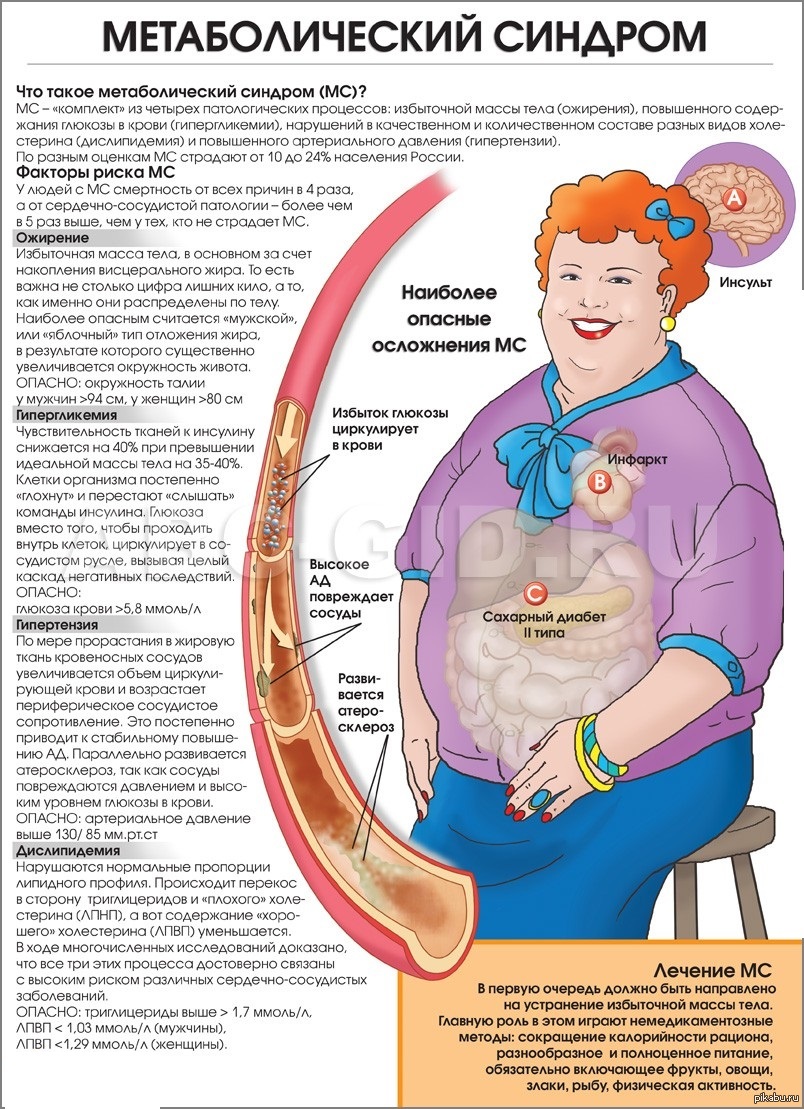 Среди сопутствующих заболеваний самый высокий уровень летальности наблюдался при заболеваниях сердечно-сосудистой системы (ССЗ) (10,5 %) и при сахарном диабете (7,3 %), несколько меньше – при хронических заболеваниях дыхательной системы (6,3 %), артериальной гипертензии (6,0 %) и онкологических заболеваниях (5,6 %) [1]. Обсуждается предположение, что между артериальной гипертензией, сахарным диабетом и коронавирусной инфекцией может существовать прямая эндокринная и метаболическая взаимосвязь, компонентом которой является ангиотензинпревращающий фермент 2 [2].
Среди сопутствующих заболеваний самый высокий уровень летальности наблюдался при заболеваниях сердечно-сосудистой системы (ССЗ) (10,5 %) и при сахарном диабете (7,3 %), несколько меньше – при хронических заболеваниях дыхательной системы (6,3 %), артериальной гипертензии (6,0 %) и онкологических заболеваниях (5,6 %) [1]. Обсуждается предположение, что между артериальной гипертензией, сахарным диабетом и коронавирусной инфекцией может существовать прямая эндокринная и метаболическая взаимосвязь, компонентом которой является ангиотензинпревращающий фермент 2 [2].
Данные ранних исследований
Данные исследований, проведенных в Китае [1] и итальянской провинции Ломбардия [3], в которых сообщалось о сопутствующих заболеваниях у пациентов с COVID-19, не содержали информации о росте и массе тела, которые необходимы для оценки массы жировой ткани с помощью расчета ИМТ. Описательное исследование, проведенное в больницах Сиэтла с выборкой из 24 пациентов (63 % – мужчины) с критически тяжелым течением COVID-19, стало одним из первых сообщений о величине ИМТ у пациентов с COVID-19. 3 пациента имели нормальный ИМТ, 7 – избыток массы тела, 13 – ожирение, относительно одного пациента не было данных. Абсолютные значения в данном исследовании слишком малы для проведения статистического анализа и однозначной трактовки результатов, но у 85 % пациентов с ожирением возникла потребность в ИВЛ, а 62 % пациентов с ожирением умерли [4]. Поскольку ССЗ и сахарный диабет сильно взаимосвязаны с повышенной массой жировой ткани [5], высокие значения ИМТ могут быть важным фактором риска тяжелого течения заболевания, в частности, пневмонии у пациентов с COVID-19.
3 пациента имели нормальный ИМТ, 7 – избыток массы тела, 13 – ожирение, относительно одного пациента не было данных. Абсолютные значения в данном исследовании слишком малы для проведения статистического анализа и однозначной трактовки результатов, но у 85 % пациентов с ожирением возникла потребность в ИВЛ, а 62 % пациентов с ожирением умерли [4]. Поскольку ССЗ и сахарный диабет сильно взаимосвязаны с повышенной массой жировой ткани [5], высокие значения ИМТ могут быть важным фактором риска тяжелого течения заболевания, в частности, пневмонии у пациентов с COVID-19.
Интерес к связи ИМТ с особенностями течения COVID-19 был поддержан предварительными результатами исследований из Шэньчжэня (Китай) и Нью-Йорка (США) (данные не прошли рецензирование). Среди 383 пациентов с COVID-19 из Шэньчжэня у пациентов с избыточной массой тела риск развития тяжелой пневмонии был выше, чем у пациентов с нормальной массой тела, на 86 %, а у пациентов с ожирением – на 142 %, при учете возможных искажающих факторов [6].
Среди 4103 пациентов с COVID-19 в одном из академических медицинских учреждений Нью-Йорка ИМТ более 40 кг/м² оказался вторым по силе независимым предиктором госпитализации после преклонного возраста [7]. Кроме того, по данным небольшого исследования в университетской больнице города Лилля (Франция), в котором участвовало 124 пациента с COVID-19, необходимость проведения инвазивной ИВЛ ассоциирована с ИМТ более 35 кг/м², вне зависимости от других сопутствующих заболеваний [8]. Предполагается, что у людей с высокими значениями ИМТ высокий риск потребности в респираторной поддержке связан с нарушениями дыхательной механики, повышенным сопротивлением дыхательных путей и нарушением газообмена, а также с другими патофизиологическими особенностями ожирения, например со слабостью дыхательных мышц и малыми дыхательными объемами [9].
Парадокс ожирения
Среди пациентов с пневмонией на фоне ожирения описан «парадокс выживаемости». Он заключается в том, что несмотря на повышенный риск развития пневмонии, трудность интубации и масочной вентиляции легких, риск смерти может оказаться ниже для пациентов с пневмонией и ожирением, чем для пациентов с пневмонией и нормальным ИМТ [10]. Возможными факторами, уравновешивающими риски, могут быть более агрессивное лечение пациентов с ожирением, их большие метаболические резервы или другие неучтенные особенности [10]. Ожирение потенциально играет значимую роль в увеличении частоты развития и степени тяжести пневмонии (возможно, и других осложнений), поэтому антропометрические данные являются важным клиническим аспектом при обследовании пациента с COVID-19.
Возможными факторами, уравновешивающими риски, могут быть более агрессивное лечение пациентов с ожирением, их большие метаболические резервы или другие неучтенные особенности [10]. Ожирение потенциально играет значимую роль в увеличении частоты развития и степени тяжести пневмонии (возможно, и других осложнений), поэтому антропометрические данные являются важным клиническим аспектом при обследовании пациента с COVID-19.
Кроме того, метаболические нарушения (включающие артериальную гипертензию, дислипидемию и гипергликемию), ассоциированные с ожирением, могут также наблюдаться у пациентов с нормальной или избыточной массой тела [5]. Предиабет, которым страдает 38 % взрослого населения США, является важным фактором риска развития ССЗ и заболеваний почек [5]. Однако остается неясным, в какой степени данные кардиометаболические факторы риска влияют на предрасположенность к развитию тяжелых заболеваний у пациента вне зависимости от его ИМТ.
Заключение
Для лучшего прогнозирования риска осложнений у пациентов с COVID-19 в дополнение к стандартной оценке параметров при госпитализации (в том числе оценке органной недостаточности по SOFA, уровня D-димера и маркеров воспалительного процесса) необходимо измерение антропометрических и метаболических показателей. К этим параметрам относятся ИМТ, окружность талии и бедер, а также уровень глюкозы и инсулина.
К этим параметрам относятся ИМТ, окружность талии и бедер, а также уровень глюкозы и инсулина.
Последние два параметра могут быть использованы для оценки инсулинорезистентности, например с помощью расчета индекса HOMA-IR. Осведомленность о наличии инсулинорезистентности у пациента важна, потому что последняя является одной из важнейших детерминант риска развития метаболических нарушений, дисфункции миокарда и риска смерти от ССЗ [5]. Данные измерения могут быть полезны как в условиях оказания первичной медицинской помощи, так и в условиях стационара для оценки риска осложненного течения заболевания у пациентов с лабораторно подтвержденным SARS-CoV-2 (Рис. 1).
закрыть менюПациенты с ожирением часто страдают дыхательными нарушениями, для которых характерно изменение механизма дыхания, повышение сопротивления дыхательных путей, нарушение газообмена, уменьшение дыхательных объемов и слабость дыхательной мускулатуры. Люди с такими нарушениями предрасположены к развитию пневмонии вследствие гиповентиляции, легочной гипертензии и сердечной недостаточности. Ожирение также ассоциировано с повышенным риском развития сахарного диабета, ССЗ и заболеваний почек – сопутствующих заболеваний, которые, как считается, повышают вероятность органной недостаточности, связанной с пневмонией. Однако, даже при отсутствии сопутствующих ожирению заболеваний, наличие артериальной гипертензии, дислипидемии, предиабета и инсулинорезистентности могут выступать предрасполагающими факторами для развития сердечно-сосудистых событий и увеличивать восприимчивость к инфекции посредством сопровождающих их состояний: атеросклероза, дисфункции миокарда и недостаточности иммунного ответа.
Ожирение также ассоциировано с повышенным риском развития сахарного диабета, ССЗ и заболеваний почек – сопутствующих заболеваний, которые, как считается, повышают вероятность органной недостаточности, связанной с пневмонией. Однако, даже при отсутствии сопутствующих ожирению заболеваний, наличие артериальной гипертензии, дислипидемии, предиабета и инсулинорезистентности могут выступать предрасполагающими факторами для развития сердечно-сосудистых событий и увеличивать восприимчивость к инфекции посредством сопровождающих их состояний: атеросклероза, дисфункции миокарда и недостаточности иммунного ответа.
В заключение, в то время как связь артериальной гипертензии, сахарного диабета и ССЗ с более тяжелым течением COVID-19 является общеизвестной, ожирение не было подробно изучено в свете данной проблемы. Ожирение является основным фактором риска для перечисленных заболеваний и, в более общем смысле, – для метаболических нарушений (таких как дислипидемия и инсулинорезистентность), а также оказывается связано с увеличением риска развития пневмонии. Измерение антропометрических и метаболических параметров необходимо для лучшей оценки риска развития осложнений у пациентов с COVID-19.
Измерение антропометрических и метаболических параметров необходимо для лучшей оценки риска развития осложнений у пациентов с COVID-19.
Список литературы
- Wu, Z. & McGoogan, J. M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in china: summary of a report of 72,314 cases from the Chinese Center for Disease Control and Prevention. JAMA 323, 1239–1242 (2020).
- Bornstein, S. R., Dalan, R., Hopkins, D., Mingrone, G. & Boehm, B. O. Endocrine and metabolic link to coronavirus infection. Nat. Rev. Endocrinol. doi.org(2020).
- Grasselli, G. et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA doi.org (2020).
- Bhatraju, P. K. et al. Covid-19 in critically ill patients in the Seattle region-Case Series. N. Engl. J. Med. doi.org (2020).
- Stefan, N., Schick, F. & Häring, H. U. Causes, characteristics, and consequences of metabolically unhealthy normal weight in humans.
 Cell Metab. 26, 292–300 (2017).
Cell Metab. 26, 292–300 (2017). - Qingxian, C. et al. Obesity and COVID-19 severity in a designated hospital in Shenzhen, China. Preprint at SSRN doi.org(2020).
- Petrilli, C. M. et al. Factors associated with hospitalization and critical illness among 4,103 patients with COVID-19 disease in New York City. Preprint at medRxiv doi.org (2020).
- Simonnet, A. et al. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity doi.org(2020).
- Murugan, A. T. & Sharma, G. Obesity and respiratory diseases. Chron. Respir. Dis. 5, 233–242 (2008).
- Nie, W. et al. Obesity survival paradox in pneumonia: a meta-analysis. BMC Med. 12, 61 (2014).
 Cell Metab. 26, 292–300 (2017).
Cell Metab. 26, 292–300 (2017).Отделение эндокринологических заболеваний Клиника Лив Улус, которое также может быть названо отделением гормональных болезней, имеет все возможности, связанные с гормональными тестами и методами радиологической визуализации, необходимыми в области эндокринологии и метаболических заболеваний. Эндокринологические пациенты находятся под наблюдением и получают комплексное лечение с помощью специализированных напрвлений, таких как эндокринная хирургия, оториноларингология, гипофизарная хирургия (нейрохирургия), ядерная медицина, «диабетическая стопа», кардиология.
Эндокринологические пациенты находятся под наблюдением и получают комплексное лечение с помощью специализированных напрвлений, таких как эндокринная хирургия, оториноларингология, гипофизарная хирургия (нейрохирургия), ядерная медицина, «диабетическая стопа», кардиология.
Отделение эндокринологии и метаболических заболеваний специализируется на гормональных и метаболических болезнях. В основном это:
• Сахарный диабет (диабет 1 типа, диабет 2 типа, гестационный диабет)
• Ожирение (ожирение)
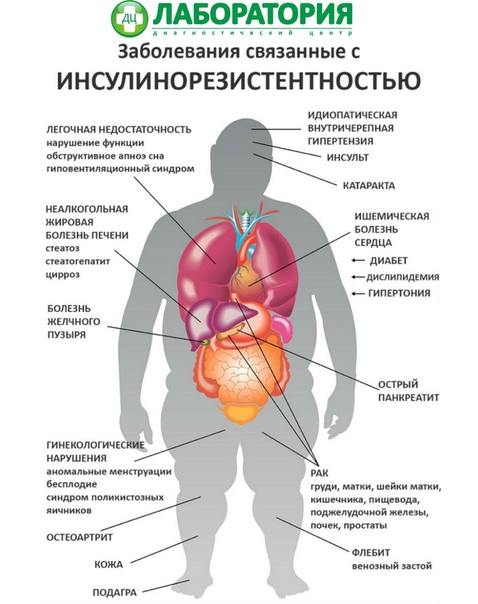
• Состояния резистентности к инсулину (метаболический синдром и синдром поликистозных яичников)
• Слабость (нервная анорексия, булимия)
• Заболевания щитовидной железы (зоб, узлы щитовидной железы, рак щитовидной железы, воспаление щитовидной железы)
• Заболевания паращитовидной железы
• Болезни гипофиза (задержка роста — низкорослость, гигантизм, акромегалии)
• Заболевания надпочечников
• Гирсутизм (оволосение по мужскому типу)
• Гормональные заболевания яичков и яичника
• Бесплодие
• Остеопороз и другие метаболические заболевания костей
• Гипертония (высокое кровяное давление)
• Заболевания обмена липидов (жиров в крови) и редкие заболевания обмена веществ.
Клиническое обслуживание предоставляется во всех этих областях эндокринологии.
Команда «диабетической стопы» В нашей клинике пациенты, страдающие диабетом, находятся под наблюдением команды опытных эндокринологов, диетологов и диабетических медсестер, а так же пациентам предоставляется обучение по диабету. В нашей поликлинике применяются такие системы технологического лечения и мониторинга, как инсулиновая помпа и постоянный мониторинг уровня глюкозы в крови. Проблемы с глазами (ретинопатия), повреждение нервов (нейропатия), а также связанные с диабетом осложнения, рассматриваются работающими в сотрудничестве опытными офтальмологами, неврологами, Раны диабетической стопы, которые являются серьезным осложнением и могут привести к потере конечности, если их не лечить быстро, исследуются и лечатся командой «диабетической стопы», которая состоит из врачей-специалистов соответствующих подразделений. В результате, наше отделение располагает всем техническим оборудованием и медицинскими услугами, необходимыми для диабетического центра.

Команда тироидологов
Команда тироидологов Клиники Лив оказывает услуги по всем процессам обследования, лечения и наблюдения заболеваний щитовидной железы, которые встречаются у каждого третьего взрослого в нашей стране. Отделение тироидологии, которому для совместной работы требуется множество отделений, состоит из отделения эндокринологии и обмена веществ, эндокринной хирургии, радиологии, патологии, ядерной медицины, офтальмологии (здоровье глаз), оториноларингологии. В отделении, куда направляются пациенты, желающие записаться на прием по поводу заболеваний щитовидной и околощитовидной железы, лечение планируется в рамках научных подходов, принятых во всем мире, после первоначальной оценки и быстрого завершения исследований командой тироидологов.Совет по тиреоидологии
Совет по тиреоидологии, в котором учавствуют все врачи соответствующих отделений, в сложных случаях с междисциплинарной точки зрения.определяет метод лечения.Метаболические заболевания | Janssen
Home › Метаболические заболеванияМетаболические заболевания
Мы стремимся к тому, чтобы наши препараты (имеющиеся и находящиеся в разработке) способствовали лечению и профилактике опасных для жизни заболеваний, которые значительно влияют на общее состояние здоровья. К ним относятся, в частности, хроническая болезнь почек и ожирение.
К ним относятся, в частности, хроническая болезнь почек и ожирение.
Хроническая болезнь почек, которая поражает почти половину пациентов с диабетом 2-го типа, повышает риск развития почечной недостаточности и сердечно-сосудистых заболеваний. Сейчас лечение заболеваний почек на последней стадии ограничивается диализом и трансплантацией почек. Эти дорогостоящие процедуры не способны надолго увеличить продолжительность жизни. Несмотря на острую потребность, уже два десятилетия не появлялось новых методов лечения диабетической болезни почек. Мы стремимся в ближайшем будущем исправить это. Изучая дополнительные преимущества нашего ингибитора натрий-зависимого переносчика глюкозы 2 типа (SGLT2), а также ряда перспективных разрабатываемых препаратов, мы приближаем появление новых методов лечения, которые помогут пациентам с хронической болезнью почек.
Почти 40% взрослого населения в США и 13% во всем мире страдают ожирением. Ожирение способно повысить риск сердечно-сосудистых заболеваний, диабета, неалкогольной жировой болезни печени и других опасных для жизни осложнений. Именно поэтому мы занимаемся поиском эффективных методов снижения веса для пациентов с ожирением, входящих в группу риска.
Именно поэтому мы занимаемся поиском эффективных методов снижения веса для пациентов с ожирением, входящих в группу риска.
Cardiovascular & Metabolism
Наследственные нарушения обмена веществ — Симптомы и причины
Обзор
Унаследованные метаболические расстройства относятся к различным типам заболеваний, вызванных генетическими дефектами — чаще всего наследуемыми от обоих родителей — которые мешают обмену веществ в организме. Эти состояния также можно назвать врожденными нарушениями обмена веществ.
Метаболизм — это сложный набор химических реакций, которые ваше тело использует для поддержания жизни, включая производство энергии.Специальные ферменты расщепляют пищу или определенные химические вещества, поэтому ваше тело может сразу же использовать их в качестве топлива или хранить их. Кроме того, определенные химические процессы расщепляют вещества, которые больше не нужны вашему организму, или вырабатывают те, которых ему не хватает.
Когда эти химические процессы не работают должным образом из-за дефицита гормонов или ферментов, возникает нарушение обмена веществ. Унаследованные метаболические нарушения делятся на разные категории, в зависимости от конкретного вещества и от того, накапливается ли оно в вредных количествах (потому что оно не может быть расщеплено), слишком мало или отсутствует.
Существуют сотни наследственных нарушений обмена веществ, вызванных различными генетическими дефектами. Примеры включают:
Некоторые нарушения обмена веществ можно диагностировать с помощью обычных скрининговых тестов, проводимых при рождении. Другие выявляются только после того, как у ребенка или взрослого проявляются симптомы заболевания.
Лечение наследственного метаболического нарушения зависит от типа и степени тяжести заболевания. Поскольку существует так много типов наследственных нарушений обмена веществ, рекомендации по лечению могут значительно различаться — от диетических ограничений до трансплантации печени.
Продукты и услуги
Показать больше продуктов от Mayo ClinicЛечение наследственных нарушений обмена веществ в клинике Мэйо
12 июля 2017 г.
Показать ссылки- Goldman L, et al., Eds. Подход к врожденным ошибкам обмена веществ. В: Медицина Гольдмана-Сесила. 25-е изд. Филадельфия, Пенсильвания.: Сондерс Эльзевьер; 2016 г. https://www.clinicalkey.com. По состоянию на 11 апреля 2017 г.
- Клигман Р.М. и др. Подход к врожденным ошибкам обмена веществ. В: Учебник педиатрии Нельсона. 20-е изд. Филадельфия, Пенсильвания: Эльзевир; 2016 г. https://www.clinicalkey.com. По состоянию на 11 апреля 2017 г.
- Саттон VR. Врожденные нарушения обмена веществ: Классификация. https://www.uptodate.com/home. По состоянию на 11 апреля 2017 г.
- Саттон VR. Врожденные нарушения обмена веществ: определение конкретного заболевания. https://www.uptodate.com/home. По состоянию на 11 апреля 2017 г.
- Саттон VR. Врожденные нарушения обмена веществ: эпидемиология, патогенез и клинические особенности. https://www.uptodate.com/home. По состоянию на 11 апреля 2017 г.
- Коричневый AY. Allscripts EPSi. Клиника Мэйо, Рочестер, Миннесота, 11 апреля 2017 г.
- Lanpher BC (заключение экспертов). Клиника Мэйо, Рочестер, Миннесота, 16 мая 2017 г.
 https://www.uptodate.com/home. По состоянию на 11 апреля 2017 г.
https://www.uptodate.com/home. По состоянию на 11 апреля 2017 г.Связанные
Продукты и услуги
Показать больше продуктов и услуг Mayo ClinicНаследственные нарушения обмена веществ
Семейная гиперхолестеринемия — Симптомы и причины
Обзор
Семейная гиперхолестеринемия влияет на то, как организм перерабатывает холестерин. В результате люди с семейной гиперхолестеринемией имеют более высокий риск сердечных заболеваний и более высокий риск раннего сердечного приступа.
В результате люди с семейной гиперхолестеринемией имеют более высокий риск сердечных заболеваний и более высокий риск раннего сердечного приступа.
Ген, вызывающий семейную гиперхолестеринемию, передается по наследству. Состояние присутствует с рождения. Лечение, включая лекарства и здоровый образ жизни, может помочь снизить риски.
Симптомы
Высокий холестерин — распространенное заболевание, но часто это результат нездорового образа жизни, поэтому его можно предотвратить и вылечить.При семейной гиперхолестеринемии риск повышенного холестерина у человека выше, потому что дефект (мутация) в гене изменяет то, как организм перерабатывает холестерин. Эта мутация не позволяет организму удалять из крови холестерин липопротеинов низкой плотности (ЛПНП), «плохой» холестерин. В результате бляшки могут вызывать сужение и затвердевание артерий, повышая риск сердечных заболеваний. Генетическое тестирование может определить, есть ли у вас эта мутация.
Эти генные мутации передаются от родителей к ребенку.Чтобы иметь это заболевание, дети должны унаследовать измененную копию гена от одного из родителей. У большинства людей с семейной гиперхолестеринемией есть один пораженный ген и один нормальный ген. В редких случаях человек наследует пораженную копию от обоих родителей, что может привести к более тяжелой форме заболевания.
Причины
Семейная гиперхолестеринемия вызывается геном, который передается от одного или обоих родителей. Люди, страдающие этим заболеванием, рождаются с ним.Этот дефект не позволяет организму избавляться от холестерина, который может накапливаться в артериях и вызывать сердечные заболевания. Этот тип холестерина называется липопротеином низкой плотности, но также широко известен как ЛПНП или плохой холестерин. Холестерин ЛПНП может привести к сужению и сужению артерий. Это увеличивает риск сердечного приступа и сердечных заболеваний.
Факторы риска
Риск семейной гиперхолестеринемии выше, если один или оба ваших родителя имеют генный дефект, который ее вызывает. У большинства людей есть один пораженный ген. Но в редких случаях ребенок может получить пораженный ген от обоих родителей. Это может вызвать более тяжелую форму заболевания.
У большинства людей есть один пораженный ген. Но в редких случаях ребенок может получить пораженный ген от обоих родителей. Это может вызвать более тяжелую форму заболевания.
20 декабря 2018 г.
Показать ссылки- Информация о семейной гиперхолестеринемии. Национальный институт исследования генома человека. http://www.genome.gov.По состоянию на 27 января 2016 г.
- Семейная гиперхолестеринемия. MedlinePlus. https://www.nlm.nih.gov/medlineplus. По состоянию на 3 февраля 2016 г.
- Hartgers ML, et al. Новые подходы к выявлению и лечению семейной гиперхолестеринемии. Текущие кардиологические отчеты. 2015: 17: 109.
- Pagon RA, et al., Eds. Семейная гиперхолестеринемия. В: GeneReviews. Сиэтл, Вашингтон: Университет Сиэтла, Вашингтон; 1993-2016 гг. http://www.ncbi.nlm.nih.gov/books/NBK1116. По состоянию на 6 января 2016 г.
- Национальная медицинская библиотека. Гиперхолестеринемия. Домашний справочник по генетике. http://ghr.nlm.nih.gov/condition/hypercholesterolemia. По состоянию на 6 января 2016 г.
- Интегрированное руководство по сердечно-сосудистым заболеваниям и снижению риска у детей и подростков. Бетесда, штат Мэриленд: Национальный институт сердца, легких и крови. http://www.nhlbi.nih.gov/guidelines/cvd_ped/index.htm. По состоянию на 7 декабря 2015 г.
- Группа экспертов по комплексным руководящим принципам по сердечно-сосудистым заболеваниям и снижению риска у детей и подростков: Сводный отчет.Национальный институт сердца, легких и крови. http://www.nhlbi.nih.gov. Доступ 5 января 2016 г.
- Rosenson RS, et al. Наследственные нарушения обмена холестерина ЛПНП. http://www.uptodate.com/home. По состоянию на 6 января 2016 г.
- Робинсон Дж. Управление семейной гиперхолестеринемией: обзор рекомендаций Экспертной группы Национальной липидной ассоциации по семейной гиперхолестеринемии. Журнал управляемой аптеки. 2013; 19: 139. http://www.amcp.org/WorkArea/DownloadAsset.aspx? id = 16222. По состоянию на 15 февраля 2016 г.
- Дислипидемия у детей: управление. http://www.uptodate.com/home. По состоянию на 15 января 2016 г.
- Goldman L, et al., Eds. Нарушения липидного обмена. В: Медицина Гольдмана-Сесила. 25-е изд. Филадельфия, Пенсильвания: Сондерс Эльзевьер; 2016. http://www.clinicalkey.com. Доступ 13 января 2016 г.
- Медикаментозная терапия холестерина. Американская Ассоциация Сердца. http://www.heart.org/HEARTORG/Conditions/Cholesterol/PreventionTreatmentofHighCholesterol/Drug-Therapy-for-Cholesterol_UCM_305632_Article.jsp # .Vpkg59hIiic. По состоянию на 15 января 2016 г.

 Журнал управляемой аптеки. 2013; 19: 139. http://www.amcp.org/WorkArea/DownloadAsset.aspx? id = 16222. По состоянию на 15 февраля 2016 г.
Журнал управляемой аптеки. 2013; 19: 139. http://www.amcp.org/WorkArea/DownloadAsset.aspx? id = 16222. По состоянию на 15 февраля 2016 г.Семейная гиперхолестеринемия
Ниманна-Пика — Симптомы и причины
Обзор
Ниманна-Пика — редкое наследственное заболевание, которое влияет на способность организма метаболизировать жир (холестерин и липиды) в клетках. Эти клетки не работают и со временем умирают.Болезнь Ниманна-Пика может поражать мозг, нервы, печень, селезенку, костный мозг и, в тяжелых случаях, легкие.
Эти клетки не работают и со временем умирают.Болезнь Ниманна-Пика может поражать мозг, нервы, печень, селезенку, костный мозг и, в тяжелых случаях, легкие.
Люди с этим заболеванием испытывают симптомы, связанные с прогрессирующей потерей функций нервов, головного мозга и других органов.
Ниманна-Пика может возникнуть в любом возрасте, но в основном поражает детей. Болезнь не имеет известного лечения и иногда приводит к летальному исходу. Лечение направлено на то, чтобы помочь людям жить со своими симптомами.
Уход Niemann-Pick в клинике Mayo
Продукты и услуги
Показать другие продукты от Mayo ClinicСимптомы
Симптомы Ниманна-Пика могут включать:
- Неуклюжесть и трудности при ходьбе
- Чрезмерные сокращения мышц (дистония) или движения глаз
- Нарушения сна
- Затруднения при глотании и приеме пищи
- Рецидивирующая пневмония
Три основных типа Ниманна-Пика — это типы A, B и C. Признаки и симптомы, которые вы испытываете, зависят от типа и тяжести вашего состояния. У некоторых младенцев с типом А признаки и симптомы появляются в течение первых нескольких месяцев жизни. Люди с типом B могут не проявлять признаков в течение многих лет и имеют больше шансов дожить до взрослой жизни. Люди с типом C могут не испытывать никаких симптомов до взрослого возраста.
Признаки и симптомы, которые вы испытываете, зависят от типа и тяжести вашего состояния. У некоторых младенцев с типом А признаки и симптомы появляются в течение первых нескольких месяцев жизни. Люди с типом B могут не проявлять признаков в течение многих лет и имеют больше шансов дожить до взрослой жизни. Люди с типом C могут не испытывать никаких симптомов до взрослого возраста.
Когда обращаться к врачу
Немедленно обратитесь к врачу, если у вас или вашего ребенка появятся предупреждающие признаки Ниманна-Пика.
Причины
Ниманна-Пика вызываются мутациями в определенных генах, связанных с тем, как организм метаболизирует жир (холестерин и липиды). Мутации гена Ниманна-Пика передаются от родителей к детям по схеме, называемой аутосомно-рецессивным наследованием. Это означает, что и мать, и отец должны передать дефектную форму гена, чтобы ребенок был затронут.
Ниманна-Пика — прогрессирующая болезнь, от которой нет лекарства.Это может произойти в любом возрасте.
Типы Niemann-Pick
Типы A и B
Типы A и B вызваны отсутствием или неисправностью фермента сфингомиелиназы. Это влияет на способность организма усваивать жир (холестерин и липиды), что приводит к накоплению жира в клетках. Это вызывает дисфункцию клеток и, со временем, их гибель. Тип А встречается в основном у младенцев, у которых наблюдается тяжелое прогрессирующее заболевание головного мозга. Лекарства нет, поэтому большинство детей не доживают до своих первых нескольких лет.Тип B обычно возникает позже в детстве и не связан с первичным заболеванием головного мозга. Большинство людей, пораженных типом B, доживают до взрослого возраста.
Тип C
Тип С Ниманна-Пика — редкое наследственное заболевание. Генетические мутации этого типа вызывают накопление холестерина и других жиров в печени, селезенке или легких. В конечном итоге страдает и мозг.
В конечном итоге страдает и мозг.
Янв.25, 2018
Типы, причины, симптомы и методы лечения
Унаследованные метаболические нарушения — это генетические состояния, которые приводят к нарушениям обмена веществ. У большинства людей с наследственными метаболическими нарушениями дефектный ген приводит к дефициту ферментов. Существуют сотни различных генетических нарушений обмена веществ, симптомы, методы лечения и прогнозы которых сильно различаются.
Что такое метаболизм?
Метаболизм — это все химические реакции, происходящие в организме для преобразования или использования энергии.Вот несколько основных примеров метаболизма:
- Расщепление углеводов, белков и жиров в пище для высвобождения энергии.
- Преобразование избыточного азота в продукты жизнедеятельности, выводимые с мочой.
- Разрушение или преобразование химических веществ в другие вещества и транспортировка их внутрь клеток.
Метаболизм — это организованная, но хаотическая линия сборки химических веществ. Сырье, полуфабрикаты и отходы постоянно используются, производятся, транспортируются и выбрасываются.«Рабочие» на конвейере — это ферменты и другие белки, которые вызывают химические реакции.
Причины наследственных нарушений обмена веществ
При большинстве наследственных метаболических нарушений один фермент либо вообще не вырабатывается организмом, либо вырабатывается в неработающей форме. Недостающий фермент подобен отсутствующему работнику на конвейере. В зависимости от функции этого фермента его отсутствие означает, что могут накапливаться токсичные химические вещества или может не производиться необходимый продукт.
Код или план производства фермента обычно содержится в паре генов. Большинство людей с наследственными нарушениями обмена веществ наследуют две дефектные копии гена — по одной от каждого родителя. Оба родителя являются «носителями» плохого гена, то есть они несут одну дефектную копию и одну нормальную копию.
У родителей нормальная копия гена компенсирует плохую копию. Уровень их ферментов обычно адекватен, поэтому у них может не быть симптомов генетического нарушения обмена веществ. Однако ребенок, унаследовавший две дефектные копии гена, не может производить достаточно эффективных ферментов, и у него развивается генетическое нарушение обмена веществ.Эта форма генетической передачи называется аутосомно-рецессивным наследованием.
Первопричиной большинства генетических нарушений обмена веществ является мутация гена, произошедшая много-много поколений назад. Мутация гена передается из поколения в поколение, обеспечивая ее сохранение.
Каждое наследственное нарушение обмена веществ довольно редко встречается в общей популяции. В совокупности наследственные нарушения обмена веществ могут поражать примерно 1 из 1000–2 500 новорожденных. У некоторых этнических групп населения, таких как евреи-ашкенази (евреи центрально-восточноевропейского происхождения), частота наследственных нарушений обмена веществ выше.
Типы наследственных нарушений обмена веществ
Выявлены сотни наследственных нарушений обмена веществ, и продолжают обнаруживаться новые. Некоторые из наиболее распространенных и важных генетических нарушений метаболизма включают:
Лизосомные нарушения накопления : Лизосомы — это пространства внутри клеток, которые расщепляют отходы метаболизма. Дефицит различных ферментов внутри лизосом может привести к накоплению токсичных веществ, вызывая нарушения обмена веществ, в том числе:
- Синдром Гурлера (аномальная структура костей и задержка развития)
- Болезнь Ниманна-Пика (у детей развивается увеличение печени, затруднения при кормлении и повреждение нервов)
- Болезнь Тея-Сакса (прогрессирующая слабость у месячного ребенка, прогрессирующая до тяжелого повреждения нервов; ребенок обычно доживает до 4-5 лет)
- Болезнь Гоше (боль в костях, увеличение печени и низкое количество тромбоцитов, часто легкая форма, у детей или взрослых)
- Болезнь Фабри (боль в конечностях в детстве, при заболеваниях почек и сердца и инсультах во взрослом возрасте; поражаются только мужчины)
- Болезнь Краббе (прогрессирующее поражение нервов, задержка развития у маленьких детей; иногда взрослые)
Галактоземия: Нарушение расщепления сахарной галактозы приводит к желтухе, рвоте и поражению печени увеличение новорожденного после грудного или искусственного вскармливания.
Болезнь мочи кленового сиропа: Дефицит фермента BCKD вызывает накопление аминокислот в организме. В результате повреждаются нервы, и моча пахнет сиропом.
Фенилкетонурия (PKU): Дефицит фермента PAH приводит к высокому уровню фенилаланина в крови. Если заболевание не распознается, наступает умственная отсталость.
Болезни накопления гликогена: Проблемы с накоплением сахара приводят к низкому уровню сахара в крови, болям в мышцах и слабости.
Митохондриальные нарушения: Проблемы внутри митохондрий, электростанций клеток, приводят к повреждению мышц.
Атаксия Фридрейха: Проблемы, связанные с белком под названием фратаксин, вызывают повреждение нервов и часто проблемы с сердцем. Неспособность ходить обычно наступает в молодом возрасте.
Пероксисомные расстройства: Подобно лизосомам, пероксисомы представляют собой крошечные пространства, заполненные ферментами внутри клеток. Плохая функция ферментов внутри пероксисом может привести к накоплению токсичных продуктов метаболизма.К пероксисомным расстройствам относятся:
- Синдром Зеллвегера (аномальные черты лица, увеличенная печень и повреждение нервов у младенцев)
- Адренолейкодистрофия (симптомы повреждения нервов могут развиться в детстве или в раннем взрослом возрасте в зависимости от формы).
Метаболизм металлов нарушения: Уровни микроэлементов в крови контролируются специальными белками. Унаследованные метаболические нарушения могут привести к нарушению функции белков и токсическому накоплению металлов в организме:
Органические ацидемии: метилмалоновая ацидемия и пропионовая ацидемия.
Нарушения цикла мочевины: Дефицит орнитин-транскарбамилазы и цитруллинемия
Симптомы наследственных нарушений обмена веществ
Симптомы генетических нарушений обмена веществ широко варьируются в зависимости от имеющихся проблем метаболизма. Некоторые симптомы наследственных нарушений обмена веществ включают:
Симптомы могут возникать внезапно или медленно прогрессировать. Симптомы могут быть вызваны продуктами питания, лекарствами, обезвоживанием, незначительными заболеваниями или другими факторами. Симптомы появляются в течение нескольких недель после рождения при многих заболеваниях.Симптомы других наследственных метаболических нарушений могут развиться через годы.
Диагностика наследственных нарушений обмена веществ
Наследственные нарушения обмена веществ присутствуют при рождении, а некоторые выявляются при плановом обследовании. Во всех 50 штатах проводится скрининг новорожденных на фенилкетонурию (ФКУ). Большинство штатов также проверяют новорожденных на галактоземию. Однако никаких государственных тестов на выявление всех известных наследственных нарушений обмена веществ у младенцев не проводилось.
Усовершенствованная технология тестирования побуждает многие штаты расширять скрининг новорожденных на генетические нарушения обмена веществ.Национальный центр скрининга новорожденных и генетических ресурсов предоставляет информацию о методах скрининга в каждом штате.
Если наследственное нарушение обмена веществ не обнаруживается при рождении, его часто не диагностируют до появления симптомов. При появлении симптомов доступны специальные анализы крови или ДНК для диагностики большинства генетических нарушений обмена веществ. Направление в специализированный центр (обычно при университете) увеличивает шансы на постановку правильного диагноза.
Лечение наследственных нарушений обмена веществ
Доступны ограниченные методы лечения наследственных нарушений обмена веществ.Существенный генетический дефект, вызывающий это состояние, не может быть исправлен с помощью современных технологий. Вместо этого лечение пытается обойти проблему с метаболизмом.
Лечение генетических нарушений метаболизма основывается на нескольких общих принципах:
- Уменьшите или исключите потребление любых продуктов питания или лекарств, которые не могут метаболизироваться должным образом.
- Замените фермент или другое химическое вещество, которое отсутствует или неактивно, чтобы восстановить метаболизм до максимально близкого к норме.
- Удаляет токсичные продукты метаболизма, которые накапливаются из-за нарушения обмена веществ.
Лечение может включать такие меры, как:
- Специальные диеты, исключающие определенные питательные вещества
- Прием заменителей ферментов или других добавок, поддерживающих метаболизм
- Обработка крови химическими веществами для детоксикации опасных побочных продуктов метаболизма
По возможности , человек с наследственным нарушением обмена веществ должен получить помощь в медицинском центре, имеющем опыт лечения этих редких состояний.
Дети и взрослые с наследственными нарушениями обмена веществ могут серьезно заболеть, требуя госпитализации, а иногда и жизнеобеспечения.Лечение во время этих эпизодов направлено на оказание неотложной помощи и улучшение функции органов.
Нарушения обмена веществ | Информационный центр по генетическим и редким заболеваниям (GARD) — программа NCATS
Дефицит 17-альфа-гидроксилазыдефицит 17-бета-гидроксистероиддегидрогеназы 3
18 Недостаток гидроксилазы
2-гидроксиглутаровая ацидурия
Дефицит 2-метилбутирил-КоА дегидрогеназы
Дефицит 3-альфа гидроксиацил-КоА дегидрогеназы
3-гидроксиизомасляная ацидурия
Дефицит 3-метилкротонил-КоА-карбоксилазы
Дефицит 3-метилглутаконил-КоА гидратазы (дефект AUH)
дефицит 5-оксопролиназы
дефицит 6-пирувоилтетрагидроптеринсинтазы
Метаболический синдром абдоминального ожирения
Абеталипопротеинемия
Акаталаземия
Ацерулоплазминемия
Дефицит ацетил-КоА-ацетилтрансферазы 2
Дефицит ацетил-карнитина
Энтеропатический акродерматит
Акромегалия
Острая перемежающаяся порфирия
Дефицит аденинфосфорибозилтрансферазы
Дефицит аденозиндезаминазы
Дефицит аденозинмонофосфатдезаминазы 1
Дефицит аденилосукциназы
Адреномиелоневропатия
Болезнь тела взрослых, связанная с полиглюкозаном
Синдром глухоты альбинизма
Альбинизм глазная нейросенсорная глухота с поздним началом
ALG1-CDG (CDG-Ik)
ALG11-CDG (CDG-Ip)
ALG12-CDG (CDG-Ig)
ALG13-CDG
ALG2-CDG (CDG-Ii)
ALG3-CDG (CDG-Id)
ALG6-CDG (CDG-Ic)
ALG8-CDG (CDG-Ih)
ALG9-CDG (CDG-IL)
Алькаптонурия
Синдром Альперса
Дефицит антитрипсина альфа-1
Дефицит альфа-кетоглутаратдегидрогеназы
Альфа-маннозидоз
Дефицит аминоацилазы 1
Анемия, вызванная недостаточностью аденозинтрифосфатазы
Анемия, сидеробластическая и спиноцеребеллярная атаксия
Очевидный избыток минералокортикоидов
Дефицит аргиназы
Аргинино-янтарная ацидурия
Дефицит декарбоксилазы ароматических L-аминокислот
Синдром холестаза дисфункции почек артрогрипоз
Синдром искусств
Аспартилгликозаминурия
Атаксия с глазодвигательной апраксией 1 типа
Атаксия с дефицитом витамина Е
Атрансферринемия
Атипичная болезнь Гоше, вызванная дефицитом сапозина C — см. Болезнь Гоше
Аутоиммунный полигландулярный синдром типа 2
Аутосомно-доминантный нейрональный цероидный липофусциноз 4B
Аутосомно-доминантная атрофия зрительного нерва и катаракта
Аутосомно-доминантная атрофия зрительного нерва плюс синдром
Аутосомная эритропоэтическая протопорфирия
Аутосомно-рецессивный нейрональный цероидный липофусциноз 4A — см. Цероидный липофусциноз взрослых нейронов
Аутосомно-рецессивная спастическая атаксия 4
Аутосомно-рецессивная спиноцеребеллярная атаксия 9
B4GALT1-CDG (CDG-IId)
Сидероз банту
Синдром Барта
Синдром Барттера
Синдром Барттера антенатальный тип 1
Синдром Барттера антенатальный тип 2
Синдром Барттера тип 3
Синдром Барттера тип 4
Дефицит бета-кетотиолазы
Биотин-тиамин-зависимая болезнь базальных ганглиев
Дефицит биотинидазы
Синдром Бьорнстада
Синдром синего подгузника
Дефицит карбамоилфосфатсинтетазы 1
Дефицит карнитин пальмитоил трансферазы 1A
Дефицит карнитин-ацилкарнитинтранслоказы
Карнозинемия
Центральный несахарный диабет
Церебральная недостаточность фолиевой кислоты
Церебротехнический ксантоматоз
Цероидный липофусциноз нейрональный 1
Синдром Чанарина-Дорфмана
Синдром Чедиака-Хигаши
ДЕТСКИЙ синдром
Гипофосфатазия в детстве
Болезнь накопления холестерилового эфира
Хондрокальциноз 1
Хондрокальциноз 2
Хондрокальциноз, вызванный отложением кристаллов апатита
Chondrodysplasia punctata 1, Х-сцепленная рецессивная
Хроническая прогрессирующая наружная офтальмоплегия
Хиломикронная ретенционная болезнь
Дефект транспорта цитруллина
Цитруллинемия II типа
COG1-CDG (CDG-IIg)
COG4-CDG (CDG-IIj)
COG5-CDG (CDG-IIi)
COG7-CDG (CDG-IIe)
COG8-CDG (CDG-IIh)
Недостаток комбинированного окислительного фосфорилирования 16
Врожденный дефект синтеза желчных кислот 1-го типа
Врожденный дефект синтеза желчных кислот 2-го типа
Врожденное нарушение гликозилирования I / IIX типа
Врожденная дизеритропоэтическая анемия 2 типа
Врожденная эритропоэтическая порфирия
Врожденная лактазная недостаточность
,00 Врожденная мышечная дистрофия-дистрогликанопатия с умственной отсталостью или без нее (тип B)
Дефицит меди, доброкачественный семейный характер
CoQ-чувствительный дефицит OXPHOS
Синдром Криглера-Наджара, тип 1
Синдром Криглера-Наджара тип 2
Цистиноз
Дефицит цитохром с оксидазы
D-2-гидроксиглутаровая ацидурия
D-бифункциональная белковая недостаточность
D-глицерикацидемия
Болезнь Данона
DCMA синдром
DDOST-CDG (CDG-Ir)
Глухота, дистония и церебральная гипомиелинизация
Дентаторубрально-паллидолуйзийская атрофия
Десмостеролоз
Анемия Даймонда-Блэкфана
Дикарбоновая аминоацидурия
Дефицит дигидролипоамиддегидрогеназы
Дефицит дигидроптеридинредуктазы
Дефицит дигидропиримидиназы
Дефицит дигидропиримидиндегидрогеназы — Не редкое заболевание
Несахарный дипсогенный диабет
ДОЛК-CDG (CDG-Im)
Допа-зависимая дистония
Дефицит дофамин-бета-гидроксилазы
Болезнь Даулинга-Дегоса
DPAGT1-CDG (CDG-Ij)
DPM1-CDG (CDG-Ie)
DPM2-CDG
DPM3-CDG (CDG-Ио)
Синдром Дубина-Джонсона
Энцефалопатия из-за недостаточности просапозина — см. Сфинголипидоз
Эритропоэтическая уропорфирия, связанная с миелоидным злокачественным новообразованием
Этилмалоновая энцефалопатия
Болезнь Фабри
Синдром семейной хиломикронемии
Семейный дефицит ЛПВП
Семейная гипокальциурическая гиперкальциемия 1 типа
Семейная гипокальциурическая гиперкальциемия 2 типа
Семейная гипокальциурическая гиперкальциемия 3 типа
Семейный дефицит LCAT
Семейная частичная липодистрофия 2 типа
Синдром Фанкони-Бикеля
Болезнь Фарбера
Детская энцефаломиопатия со смертельным исходом
Нейродегенерация, связанная с гидроксилазой жирных кислот
Рыбий глаз
Дефицит фруктозо-1,6-бисфосфатазы
Фукозидоз
Мышечная дистрофия Фукуямы
Дефицит фумаразы
Дефицит галактокиназы
Галактосиалидоз
Дефицит трансаминазы гамма-аминомасляной кислоты
Дефицит гамма-цистатионазы
Болезнь Гоше
Болезнь Гоше — офтальмоплегия — сердечно-сосудистая кальцификация — см. Болезнь Гоше
Болезнь Гоше перинатальный летальный исход
Болезнь Гоше 1 типа
Болезнь Гоше 2 типа
Болезнь Гоше тип 3
Несахарный гестационный диабет
Синдром Гилберта — Не редкое заболевание
Синдром Гительмана
Синдром дефицита переносчика глюкозы типа 1
Мальабсорбция глюкозы-галактозы
Дефицит глутаматформиминотрансферазы
Недостаток глутамина врожденный
Глутаровая ацидемия I типа
Глутаровая ацидемия II типа
Глутаровая ацидемия III типа
Дефицит глутатионсинтетазы
Глутатионурия
Дефицит глицин-N-метилтрансферазы
Болезнь накопления гликогена 8
Болезнь накопления гликогена типа 0, печень
Болезнь накопления гликогена, тип 12
Болезнь накопления гликогена, тип 13
Болезнь накопления гликогена типа 1А
Болезнь накопления гликогена типа 1B
Болезнь накопления гликогена, тип 3
Болезнь накопления гликогена, тип 5
Болезнь накопления гликогена, тип 6
Болезнь накопления гликогена, тип 7
Гликопротеиноз
GM1 ганглиозидоз 1 типа
GM1 ганглиозидоз 2 типа
GM1 ганглиозидоз 3 типа
дефицит GM3-синтазы
Синдром ГРАЦИЛА
Дисплазия Гринберга
дефицит GTP циклогидролазы I
Дефицит гуанидиноацетатметилтрансферазы
Гиратная атрофия сосудистой оболочки и сетчатки
Синдром Хаима-Мунка
Болезнь Хартнупа
Хокинсинурия
Гемохроматоз 2 типа
Гемохроматоз 3 типа
Гемохроматоз 4 типа
Дефицит печеночной липазы
Гепатоэритропоэтическая порфирия
Наследственный амилоидоз
Наследственная копропорфирия
Наследственная мальабсорбция фолиевой кислоты
Наследственная непереносимость фруктозы
Наследственная гиперекплексия
Наследственные множественные остеохондромы
Наследственная сенсорная и вегетативная нейропатия типа 1Е
Наследственная сенсорная нейропатия 1 типа
Синдром Германского Пудлака 2
Гистидинемия
Дефицит лиазы HMG CoA
Гомокарнозиноз
Гомоцистеинемия
Гомоцистинурия, вызванная дефицитом CBS
Гомоцистинурия, вызванная дефицитом MTHFR
HSD10 болезнь
Синдром Гурлера
Синдром Херлера – Шейе
Гидроксикинуренинурия
Синдром гипер-IgD
Гипербетааланинемия
Синдром гиперкоагуляции, вызванный дефицитом гликозилфосфатидилинозитола
Гиперглицеролемия
Гиперинсулинизм из-за недостаточности глюкокиназы
Синдром гиперинсулинизма-гипераммониемии
Гиперлипидемия 3 типа
Гиперлипопротеинемия 5 типа
Гиперлизинемия
Гиперфенилаланинемия, вызванная дефицитом дегидратазы
Гиперпролинемия
Гиперпролинемия 2 типа
Гипертриптофанемия
Гиполипопротеинемия
Гипофосфатазия
I клеточная болезнь
Синдром Имерслунда-Грасбека
Иминоглицинурия
Миопатия с тельцами включения 2
Миопатия с тельцами включения 3
Инфантильная болезнь накопления свободной сиаловой кислоты — См. Болезнь накопления свободной сиаловой кислоты
Детская нейроаксональная дистрофия
Спиноцеребеллярная атаксия с младенческим началом
Дефицит инсулиноподобного фактора роста I.
Дефицит внутреннего фактора
Дефицит изобутирил-КоА дегидрогеназы
Изовалериановая ацидемия
Болезнь Канзаки
Синдром Кернса-Сейра
Болезнь Краббе атипична из-за дефицита сапозина А
L-2-гидроксиглутаровая ацидурия
L-аргинин: дефицит глицинамидинотрансферазы
Дефицит лактатдегидрогеназы А
Дефицит лактатдегидрогеназы
Латостеролоз
дефицит LCHAD
Наследственная оптическая нейропатия Лебера
Синдром Ли, франко-канадский тип
Синдром Леша Найхана
Лейцин-чувствительная гипогликемия младенчества
Лейкоэнцефалопатия — дистония — моторная нейропатия
Лейкоэнцефалопатия с поражением ствола и спинного мозга и повышением уровня лактата
Конечностно-поясная мышечная дистрофия 2I типа
Мышечная дистрофия пояснично-конечностного типа 2K — см. Мышечная дистрофия пояснично-конечностного типа
Мышечная дистрофия пояснично-конечностного типа 2М — см. Мышечная дистрофия пояснично-конечностного типа
Мышечная дистрофия пояснично-конечностного типа 2N — см. Мышечная дистрофия пояснично-конечностного типа
Мышечная дистрофия пояснично-конечностного типа 2O — см. Мышечная дистрофия пояснично-конечностного типа
Мышечная дистрофия пояснично-конечностного типа 2Т — см. Мышечная дистрофия пояса конечностей
Мышечная дистрофия пояснично-конечностного типа, тип 2С
Комбинированная недостаточность липазы
Дефицит синтетазы липоевой кислоты
Липоидный протеиноз Урбаха и Вите
Окулоцереброренальный синдром Лоу
Непереносимость лизинурического белка
Дефицит малонил-КоА декарбоксилазы
MAN1B1-CDG
Дефицит маннозо-связывающего лектинового белка — Не редкое заболевание
Маннозидоз, бета А, лизосомальный
Гиперфенилаланинемия матери
Сахарный диабет и глухота, наследуемые по материнской линии
Дефицит ацил-кофермента А-дегидрогеназы со средней длиной цепи
Мегалобластная анемия, вызванная недостаточностью дигидрофолатредуктазы
Болезнь Менкеса
Метахроматическая лейкодистрофия
Метахроматическая лейкодистрофия, вызванная дефицитом сапозина B
Дефицит метионин аденозилтрансферазы
Дефицит метилкобаламина cbl G тип
Метилмалоновая ацидемия с гомоцистинурией типа cblC — см. Метилмалоновая ацидемия с гомоцистинурией
Метилмалоновая ацидемия с гомоцистинурией типа cblD — см. Метилмалоновая ацидемия с гомоцистинурией
Метилмалоновая ацидемия с гомоцистинурией типа cblF — см. Метилмалоновая ацидемия с гомоцистинурией
Метилмалоновая ацидемия с гомоцистинурией типа cblJ — см. Метилмалоновая ацидемия с гомоцистинурией
Метилмалоновая ацидурия, тип cblA — см. Дефицит аденозилкобаламина
Метилмалоновая ацидурия, тип cblB — см. Дефицит аденозилкобаламина
Мевалоновая ацидурия
MGAT2-CDG (CDG-IIa)
Легкая фенилкетонурия
Дефицит митохондриального комплекса I
Дефицит митохондриального комплекса II
Дефицит митохондриального комплекса III
Синдром истощения митохондриальной ДНК, энцефаломиопатическая форма с метилмалоновой ацидурией
Синдром Ли, связанный с митохондриальной ДНК
Митохондриальная энцефаломиопатия, лактоацидоз и эпизоды, подобные инсульту
Митохондриальная миопатия и сидеробластная анемия
Митохондриальная миопатия при диабете
Митохондриальная миопатия с лактоацидозом
Синдром митохондриальной нейрогастроинтестинальной энцефалопатии
Митохондриальная трифункциональная белковая недостаточность
MOGS-CDG (CDG-IIb)
Синдром Мора-Транебьерга
Дефицит кофактора молибдена
Моногенный диабет — Не редкое заболевание
Синдром Моркио B
MPDU1-CDG (CDG-If)
MPI-CDG (CDG-Ib)
Синдром истощения митохондриальной ДНК, связанный с MPV17
Муколипидоз III альфа / бета
Муколипидоз 4 типа
Мукополисахаридоз II типа
Мукополисахаридоз III типа
Мукополисахаридоз типа IIIA
Мукополисахаридоз типа IIIB
Мукополисахаридоз IIIC типа
Мукополисахаридоз типа IIID
Мукополисахаридоз типа IVA
Мукополисахаридоз VI типа
Мукополисахаридоз VII типа
Синдром множественных врожденных аномалий-гипотонии-судорог
Синдром множественных врожденных аномалий-гипотонии-судорог 2 типа
Множественная эндокринная неоплазия 2B типа
Множественная недостаточность сульфатазы
Множественный симметричный липоматоз
Мышечная болезнь глазного мозга
Мышечная дистрофия врожденная мегакониального типа
Дефицит киназы мышечной фосфорилазы
Мышечно-контрактурный синдром Элерса-Данлоса
Миоклоническая эпилепсия с рваными красными волокнами
Миоглобинурия рецидивирующая
Дефицит N ацетилтрансферазы
Недостаточность N-ацетил-альфа-D-галактозаминидазы III типа
Дефицит N-ацетилглутаматсинтазы
NBIA / DYT / PARK-PLA2G6
Адренолейкодистрофия новорожденных
Гемохроматоз новорожденных
Неонатальный внутрипеченочный холестаз, вызванный недостаточностью цитрина
Несахарный почечный диабет
Синдром Ной-Лаксовой
Нейроферритинопатия
Цероидный липофусциноз нейронов 10
Цероидный липофусциноз нейронов 2
Цероидный липофусциноз нейронов 3
Цероидный липофусциноз нейронов 5
Цероидный липофусциноз нейронов 6
Цероидный липофусциноз нейронов 7
Цероидный липофусциноз нейронов 9
Невропатия, атаксия, пигментный ретинит, синдром
Болезнь накопления нейтральных липидов с миопатией
Болезнь Ниманна-Пика тип A
Болезнь Ниманна-Пика тип B
Болезнь Ниманна-Пика тип C1
Болезнь Ниманна-Пика тип C2
Северная эпилепсия
Не указано иное Тип 3-MGA-uria
Синдром затылочного рога
Глазной альбинизм 1 типа
Глазокожный альбинизм 1 типа
Глазокожный альбинизм 1B типа
Глазокожный альбинизм 2 типа
Глазокожный альбинизм 3 типа
OPA3 дефект
Атрофия зрительного нерва 1
Дефицит орнитин-транскарбамилазы
Синдром дефицита орнитинтранслоказы
Оротовая ацидурия 1 типа
Синдром Папийона-Лефевра
Болезнь Паркинсона 9 тип
Пароксизмальная ночная гемоглобинурия
Синдром Пирсона
Pentosuria
Постоянный неонатальный сахарный диабет
Нарушения пероксисомального биогенеза
Пероксисомные расстройства — Не редкое заболевание
Синдром Перро
Синдром Петерса плюс
PGM1-CDG
Дефицит фосфоглицераткиназы
Дефицит фосфоглицератмутазы
Повышенная активность фосфорибозилпирофосфатсинтетазы
PMM2-CDG (CDG-Ia)
Понтоцеребеллярная гипоплазия 6 типа
Поздняя кожная порфирия
Первичная недостаточность карнитина
Первичная гипероксалурия 1 типа
Первичная гипероксалурия 2 типа
Первичная гипероксалурия 3 типа
Первичная гипомагниемия с вторичной гипокальциемией
Прогрессирующая наружная офтальмоплегия аутосомно-рецессивная 1
Прогрессирующий семейный внутрипеченочный холестаз 1
Прогрессирующий семейный внутрипеченочный холестаз 2 типа
Прогрессирующий семейный внутрипеченочный холестаз 3 типа
Дефицит пролидазы
Пропионовая ацидемия
Дефицит псевдохолинэстеразы
Псевдонеонатальная адренолейкодистрофия
Дефицит пуриновой нуклеозидфосфорилазы
Пикнодизостоз
Пиридоксаль-5′-фосфат-зависимая эпилепсия
Пиридоксин-зависимая эпилепсия
Дефицит пируваткарбоксилазы
Дефицит пируватдегидрогеназного комплекса
Дефицит фосфатазы пируватдегидрогеназы
Дефицит пируваткиназы
Болезнь Рефсума
Болезнь Рефсума с повышенной пипеколической ацидемией
Болезнь Рефсума, младенческая форма
Почечная глюкозурия
Почечная гипомагниемия 2
Почечная гипомагниемия-6
Почечная тубулопатия, сахарный диабет и мозжечковая атаксия из-за дупликации митохондриальной ДНК
RFT1-CDG (CDG-In)
Rhizomelic chondrodysplasia punctata type 3 — См. Rhizomelic chondrodysplasia punctata
Роторный синдром
Сахаропинурия
Болезнь Салла — см. Болезнь накопления свободной сиаловой кислоты
Саркозинемия
Синдром Шейе
Иммуно-мышечная дисплазия Шимке
Болезнь Шиндлера тип 1
Дисплазия Шнекенбекена
дефицит SCOT
Гистиоцитоз синего цвета
Синдром Сенгерса
Сенсорная атаксическая нейропатия, дизартрия и офтальмопарез
Дефицит редуктазы сепиаптерина
Тяжелый комбинированный иммунодефицит
Дефицит короткоцепочечной ацил-КоА дегидрогеназы
Сиалидоз I типа
Сиалидоз, тип II
Сиалурия, французский тип
Ситостеролемия
Синдром Шегрена-Ларссона
SLC35A1-CDG (CDG-IIf)
SLC35A2-CDG
SLC35C1-CDG (CDG-IIc)
Синдром Смита-Лемли-Опица
Спастическая параплегия 7
Спиноцеребеллярная атаксия 28
Спиноцеребеллярная атаксия, аутосомно-рецессивная 3
Спондило-реберный дизостоз 1 — См. Спондил-реберный дизостоз
. Спондило-реберный дизостоз 2 — См. Спондил-реберный дизостоз
. Спондило-реберный дизостоз 3 — См. Спондил-реберный дизостоз
. Спондило-реберный дизостоз 4 — См. Спондил-реберный дизостоз
. Спондилокостальный дизостоз 6 — См. Спондилокостальный дизостоз
Спондилодиспластический синдром Элерса-Данлоса
Спондилоэпиметафизарная дисплазия слабость суставов
Спондилоторакальный дизостоз
SRD5A3-CDG (CDG-Iq)
SSR4-CDG
Дефицит янтарной полуальдегиддегидрогеназы
Болезнь Танжера
Болезнь Тея-Сакса
Синдром мегалобластной анемии, чувствительной к тиамину
Дефицит тиопурин-S-метилтранферазы
Тиглическая ацидемия
TMEM165-CDG (CDG-IIk)
Дефицит трансальдолазы
Дефицит транскобаламина 1
Преходящий неонатальный сахарный диабет
Дефицит треалазы
Триметиламинурия
Дефицит триозофосфатизомеразы
Дефицит тирозингидроксилазы
Временная недостаточность тирозиноксидазы
Тирозинемия 1 типа
Тирозинемия 2 типа
Тирозинемия 3 типа
Нарушения цикла мочевины
Валинемия
Пестрая порфирия
дефицит VLCAD
Синдром Уокера-Варбурга
Болезнь Вильсона
Синдром Вольфрама
Болезнь Вольмана
Синдром морщинистой кожи
Х-сцепленная адренолейкодистрофия
Х-сцепленная церебральная адренолейкодистрофия
Х-сцепленная болезнь Шарко-Мари-Тута, тип 5 — См. Болезнь Шарко-Мари-Тута
Х-сцепленная недостаточность креатина
Х-сцепленная доминантная точка хондродисплазии 2
Х-сцепленная сидеробластная анемия
Ксантинурия 1 типа
Ксантинурия 2 типа
Синдром Зеллвегера
Нарушения обмена веществ — симптомы, причины, лечение
Метаболизм — это расщепление пищи на более простые компоненты: белки, углеводы (или сахара) и жиры.Нарушения обмена веществ возникают, когда эти нормальные процессы нарушаются. Нарушения обмена веществ могут передаваться по наследству, и в этом случае они также известны как врожденные нарушения обмена веществ, или они могут быть приобретены в течение вашей жизни. Существует множество нарушений обмена веществ, и они распространены в Соединенных Штатах. Например, диабет — это нарушение обмена веществ, которым страдают около 26 миллионов американцев (Источник: CDC).
Фенилкетонурия является примером наследственного метаболического нарушения, характеризующегося неспособностью расщеплять один из строительных блоков белка, аминокислоту фенилаланин.Диабет I типа, заболевание, при котором поджелудочная железа не вырабатывает достаточно инсулина для поддержания сбалансированного уровня сахара в крови, представляет собой нарушение метаболизма сахара. Примером нарушения обмена веществ, влияющего на метаболизм жиров, является болезнь Гоше, которая характеризуется недостатком фермента глюкоцереброзидазы. Нарушения обмена веществ также могут быть осложнениями тяжелых заболеваний или состояний, включая печеночную или дыхательную недостаточность, рак, хроническое обструктивное заболевание легких (ХОБЛ, включая эмфизему и хронический бронхит) и ВИЧ / СПИД.
Огромные успехи были достигнуты в распознавании и лечении нарушений обмена веществ. Иногда возникают очень сложные пути, которые приводят к нарушению обмена веществ. В других случаях ответственность лежит исключительно на одной крохотной ошибке в ДНК человека. Эти открытия позволили ученым разработать необычные методы лечения больных, и темпы открытий продолжают ускоряться.
Симптомы метаболических нарушений различаются у разных людей и в зависимости от типа заболевания.Некоторые нарушения обмена веществ приводят к легким симптомам, которые можно контролировать с помощью лекарств и изменения образа жизни, в то время как другие могут вызывать серьезные и опасные для жизни симптомы, такие как проблемы с дыханием, судороги и органная недостаточность. Некоторые наследственные метаболические нарушения могут потребовать длительного приема пищевых добавок и лечения, в то время как метаболические нарушения, возникающие в результате другого заболевания или состояния, часто проходят после лечения основного состояния.
Немедленно обратитесь за медицинской помощью (звоните 911) в случае серьезных симптомов, таких как сильное затрудненное дыхание; синеватый оттенок губ или ногтей; захват; и изменение уровня сознания или бдительности, например, потеря сознания или отсутствие реакции.
Немедленно обратитесь за медицинской помощью , если вы лечитесь от нарушения обмена веществ, но легкие симптомы повторяются или сохраняются.
метаболических нарушений | Институт Кеннеди Кригера
Генетические метаболические заболевания — это врожденные химические ошибки организма, которые влияют на способ усвоения пищи, выработку энергии и рост тканей.
Большинство метаболических нарушений вызвано генетической недостаточностью фермента, необходимого для преобразования одного химического вещества в другое.Например, фенилкетонурия или «PKU» вызывается дефицитом фермента фенилаланингидроксилазы, который превращает диетическую аминокислоту фенилаланин в другую аминокислоту, тирозин. Дефицит фенилаланингидроксилазы приводит к накоплению токсичного уровня фенилаланина и дефициту тирозина, которые повреждают развивающийся мозг и вызывают серьезные умственные нарушения. Другие побочные эффекты метаболических заболеваний включают судороги, двигательные нарушения, плохой рост, мышечную слабость, непереносимость голодания и несоразмерное заболевание с простыми детскими инфекциями или прививками.
Некоторые метаболические заболевания проявляются в первые несколько дней жизни, тогда как другие требуют стресса, такого как лихорадка или голодание во время болезни, чтобы проявиться. Наиболее серьезные метаболические заболевания могут быть смертельными, если их не лечить сразу после рождения, в то время как другие могут вызывать только очень медленные травмы или приводить к разрушительному метаболическому кризису только один раз в жизни. Хотя каждое нарушение обмена веществ в отдельности встречается редко, известно более 1300 заболеваний обмена веществ, и в совокупности они представляют собой серьезную причину болезней и инвалидности у детей.
Хотя все государственные программы скрининга новорожденных проверяют наличие метаболических заболеваний, в большинстве штатов проверяется менее 10 наиболее распространенных из них. Специализированные лабораторные исследования, доступные только в нескольких крупных педиатрических центрах, таких как Институт Кеннеди Кригера, необходимы для диагностики большинства метаболических заболеваний.
Примеры, подмножества и синонимы для метаболических нарушений — врожденные ошибки метаболизма:
- Нарушения обмена аминокислот (аминоацидемии)
- Болезнь мочи кленового сиропа (MSUD)
- Гомоцистинурия
- Нарушения обмена органических кислот (органические ацидурии, органические ацидемии)
- Метилмалоновая ацидурия
- 3-метилглутаконовая ацидурия — синдром Барта
- Глутаровая ацидурия
- 2-гидроксиглутаровая ацидурия — формы D и L
- Нарушения бета-окисления жирных кислот
- Дефицит MCAD
- LCHAD, дефицит VLCAD
- Нарушения липидного обмена (нарушения накопления липидов)
- ганглиозидозы
- GM1 Ганглиозидоз
- Болезнь Тея-Сакса
- Болезнь Сандхоффа
- Сфинголипидозы
- Болезнь Фабри
- Болезнь Гоше
- Болезнь Ниманна-Пика
- Болезнь Краббе
- Муколипидозы
- Мукополисахаридозы
- ганглиозидозы
- Митохондриальные заболевания
- Митохондриальные кардиомиопатии
- Болезнь Ли
- МЕЛАС, МЕРРФ, НАРП
- Синдром Барта
- Пероксисомальные заболевания
- Синдром Зеллвегера (цереброгепаторенальный синдром)
- Х-сцепленная адренолейкодистрофия
- Болезнь Рефсума