Составь схему обмена веществ у растений
Здравствуйте нужна презентация по био «Заболевания которые передаются половым путём» я решила выбрать сифилис Должно быть максимум 10 слайдов и доста … точно текста даю 80 баллов
допоможіть 4 завдання біологія 6 клас
У ході досліду листок рослини змастили вазеліном. Попри те, що листок добре освітлювався Сонцем, органічні речовини ( глюкоза або крохмаль) у ньому не … утворився. Поясни цей факт. Даю 20 балов!!!
1. Органи слуху риб представлені вухом А зовнішнім Б середнім в внутрішнім г внутрішнім і середнім
Задание 1. )16 На рисунке I изображена схема строения нефрона. Укаси структуру нейрона V Петля Генле, промаксимальный каналец, клубочки, собирательная … трубочка,дистальный каналец, капилляры, 1- 2- 3 4- 5 — 6- (6) Укажите функцию нефрона. 316 Помогите даю 50 балов !!!!!
Очень нужна помощь! Нормальные в отношении зрения женщина и мужчина имеют сына, страдающего дальтонизмом, и двух дочерей с нормальным зрением.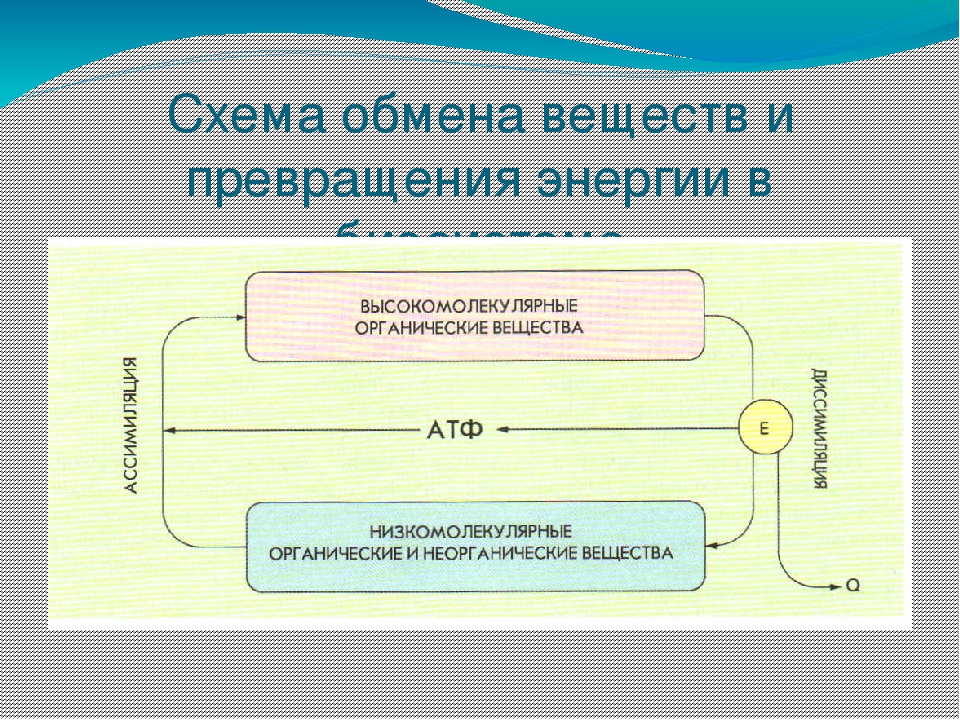
ДАЮ 100 БАЛЛОВ ПРАКТИЧЕСКАЯ РАБОТА «ТКАНИ РАСТЕНИЙ»
ЦЕЛЬ РАБОТЫ: изучить особенности растительных тканей
ХОД РАБОТЫ:
КАРТИНКИ К ПРАКТИЧЕСКОЙ ВЫ СМОЖЕТ
… Е НАЙТИ НА ЭТОМ САЙТЕ
https://infourok.ru/prakticheskaya-rabota-tkani-rasteniy-klass-2132224.html
1. Перед вами рисунок с изображением образовательной ткани растения.
А)Как вы думаете, в каких частях растения клеток образовательной ткани содержится наибольшее количество?
Б) В чем заключается функция образовательной ткани?
2. Перед вами покровная ткань растения.
А) Как вы думаете, какие структуры покровной ткани обозначены стрелками и какую важную роль они выполняют в растительном организме?
Б) Что такое чечевички и какое значение они имеют в жизни растения?
3. Перед вами основная или фотосинтезирующая ткань растения.
А) Какие важные органоиды есть в клетках этой ткани?
Б) Какое значение для жизни растения имеет фотосинтезирующая ткань?
В) Какое значение для всего органического мира имеет процесс, протекающий в клетках основной ткани растений?
4.
каковы функции стебля
Очень нужна помощь! Нормальные в отношении зрения женщина и мужчина имеют сына, страдающего дальтонизмом, и двух дочерей с нормальным зрением. Жена сы … на и мужья дочерей имеют нормальное зрение. Какова вероятность рождения в их семьях детей, страдающих дальтонизмом?
сочинение на тему: удобрения применяемые в Рязанской области.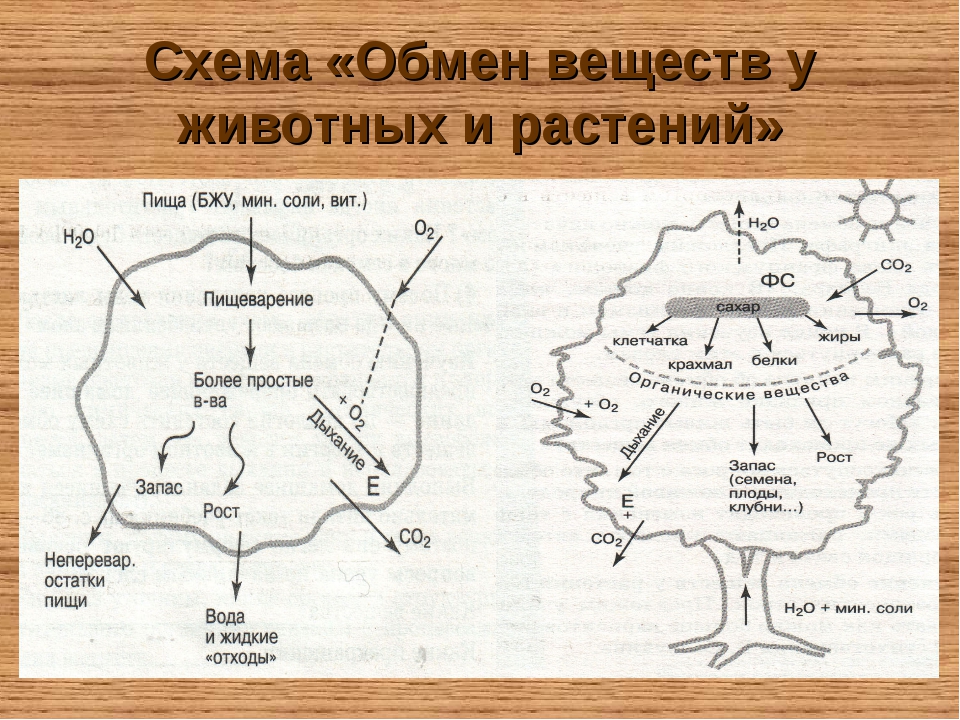 Объем: не более 1,5 листов е
Объем: не более 1,5 листов е
Препараты для нормализации обмена веществ
{{/if}} {{each list}} ${this} {{if isGorzdrav}}Удалить
{{/if}} {{/each}} {{/if}}Заказать таблетки для ускорения метаболизма
На интернет-ресурсе можно ознакомиться с инструкциями к препаратам, подобрать дешевые или дорогие аналоги по действующему веществу, изучить отзывы других заказчиков.
Применение препаратов для ускорения метаболизма
Метаболизм — важнейшая функция, комплекс энергетических и биохимических процессов, способствующих усвоению питательных веществ и расходованию их на нужды организма, удовлетворение его потребностей в энергетических и пластических веществах.1 О том, что скорость обменных реакций снизилась и необходима нормализация метаболических процессов, можно понять по признакам:
- быстрый набор лишнего веса;
- отеки лица, конечностей;
- ухудшение состояния кожных покровов, волос;
- высокая утомляемость.
Если нарушенный метаболизм спровоцировал ожирение, бесполезно покупать рядовые средства для похудения. В первую очередь, нужно привести в норму обменные процессы. Только тогда липолиз активируется. Помогут в этом современные препараты для ускорения метаболизма. Подбирать их нужно с помощью квалифицированного врача.
Формы выпуска
В каталоге сайта «Горздрав» представлены высокоэффективные лекарства для улучшения обмена веществ, выпущенные в разных формах:
- таблетки;
- кремы;
- капсулы;
- пластыри;
- сушеные травы.
Для кого
Богатый выбор препаратов для обмена веществ облегчает выбор, позволяет приобретать медикаменты, которые имеют минимальное количество побочных эффектов и хорошо переносятся больным. В наших аптеках можно заказать лекарства:
- для взрослых;
- для детей;
- лиц, с нарушениями в работе эндокринной системы.
Противопоказания
При выборе продуктов, направленных на повышение скорости протекания обменных процессов, нужно внимательно изучать противопоказания и побочные действия. Чаще всего производители указывают, что их препараты нельзя использовать:
- во время беременности;
- детям до определенного возраста;
- в период грудного вскармливания;
- при аллергии на любое из соединений состава.


Сертификаты
Некоторые сертификаты товаров, представленных в нашем каталоге.
ПЕРЕД ПРИМЕНЕНИЕМ ПРЕПАРАТОВ НЕОБХОДИМО ОЗНАКОМИТЬСЯ С ИНСТРУКЦИЕЙ ПО ПРИМЕНЕНИЮ ИЛИ ПРОКОНСУЛЬТИРОВАТЬСЯ СО СПЕЦИАЛИСТОМ.
Список литературы:
- [1] Граник В. Г. «Метаболизм эндогенных соединений», М., «Вузовская книга», 2006, 528 с.
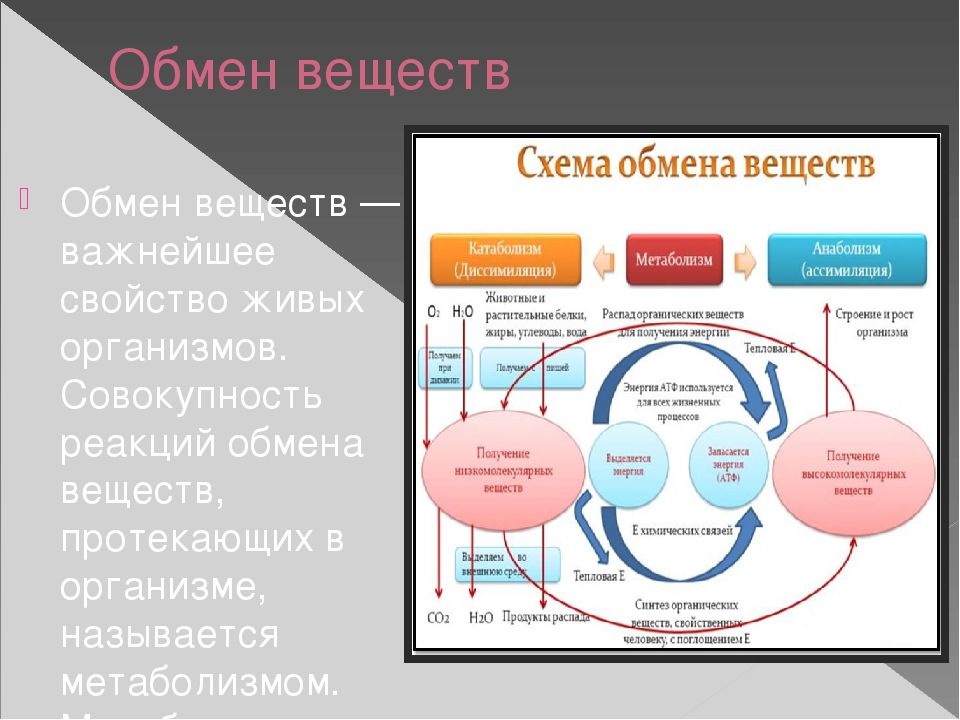
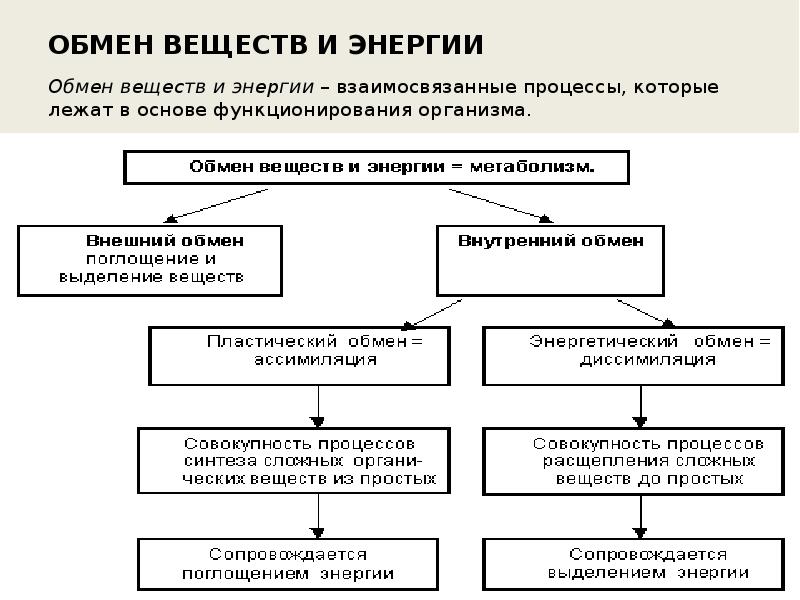
Тема урока: «Обмен веществ»
Цели урока:
- Сформулировать и расширить знания о видах обмена веществ и энергии.
- Научить школьников различать составляющие обмена веществ (ассимиляция и диссимиляция) и доказать, что эти процессы протекают параллельно.
- Выявить этапы обмена веществ и его роль в жизнедеятельности. Повторить строение и функции органических веществ.
ХОД УРОКА
1. Актуализация знаний учащихся (фронтальный опрос)
2. Изучение нового материала (объяснение
учителя)
Изучение нового материала (объяснение
учителя)
а) Объяснение понятия обмена веществ.
Учитель сравнивает процесс обмена веществ в организме с круговоротом веществ в природе. В тетрадь записываются определения:
- обмен веществ,
- пластический обмен,
- энергетический обмен,
- гомеостаз.
Объяснения учителя сопровождаются построением опорно-логической схемы на доске (Схема 1). Данную схему можно показать, используя ТСО.
Схема 1
б) Классификация видов обмена веществ по агрегатному состоянию.
Учитель просит вспомнить школьников, какими веществами может обмениваться организм с окружающей средой и на основе ответов учащихся составляется классификация:
1. Обмен газов.
2. Обмен воды и водных растворов.
3.Обмен органических веществ, включающих в себя белки, жиры, углеводы.

Более подробно вспоминают обмен органических веществ, опираясь на графическое изображение материала. Схему 2 учитель может заполнять вместе с учащимися, при этом школьники вспоминают строение органических веществ и их функции.
Схема 2
Обмен органических веществ
в) Выявление взаимосвязи органических веществ с использованием схемы 3:
Схема 3
Учащиеся, опираясь на свои предыдущие знания,
используя схему, предложенную учителем, делают
вывод о наличии взаимосвязи между органическими
веществами в организме.
После работы со схемой учащимися под
руководством педагога суммируются знания об
обмене веществ.
3. Закрепление знаний
Учитель предлагает ученикам объяснить схему обмена веществ с учетом приобретенных на уроке знаний.
1. Высокомолекулярные органические вещества
(белки, жиры, углеводы).
2. Низкомолекулярные вещества (аминокислоты,
жирные кислоты, глицерин, моносахариды).
3. АТФ.
4. Подведение итогов урока
Ответ §10. Обмен веществ и энергии
РАЗМЕЩЕНИЕ85) Сформулируйте и запишите определение
Ответ: Обмен веществ – это получение организмом из внешней среды нужных веществ и удаление из организма во внешнюю среду ненужных веществ.
86) Каково значение обмена веществ для живого организма?
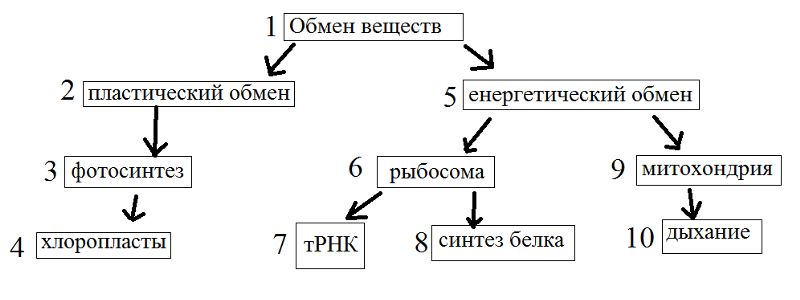
87) Какие процессы происходят в хлоропластах и митохондриях клетки?
-
Ответ: В хлоропластах – синтез органических веществ, из неорганических на свету (световой день), при использовании воды и углекислого газа.
Побочный продукт – кислород (О2).В митохондриях – происходит распад органических веществ и синтез энергии.
 Побочный продукт – кислород (О2).
Побочный продукт – кислород (О2).
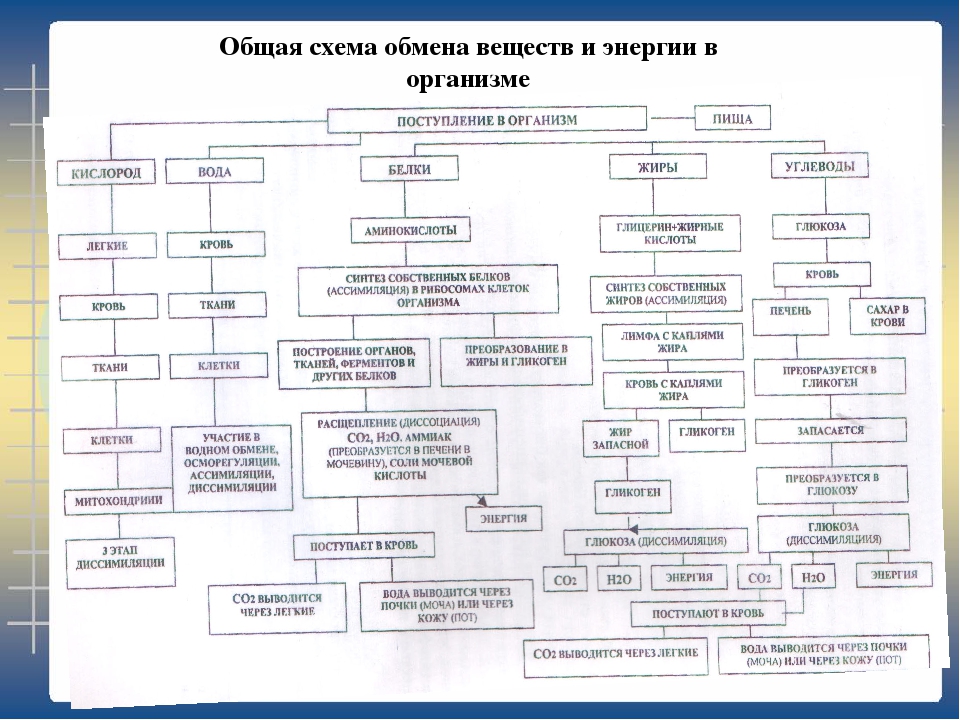
88) Заполните схему «Обмен веществ у животных»
-
Ответ:
↓Вещества, образовавшиеся в результате обмена веществ:
Вода
Углекислый газ
Продукты распада
↓Вещества, поступающие в организм:
Кислород
Белки
Жиры
Углеводы
Вода
Минеральные соли
Процессы, происходящие при обмене веществ:
Процессы жизнедеятельности
89) Заполните схему «Обмен веществ у растений»
-
Ответ:
↓Вещества, поступающие в организм:
Кислород
Углекислый газ
Свет
Вода, с растворенными веществами
↓Вещества, образовавшиеся в результате обмена веществ:
Углекислый газ
Кислород
Поры воды
Процессы, происходящие при обмене веществ:
Дыхание
Фотосинтез
90) Дайте определения
-
Ответ: Теплокровные животные – это животные, температура тела которых не зависит от температуры окружающей среды.
Холоднокровные животные – это животные, температура тела которых зависит от температуры окружающей среды.
91) Приведите примеры животных, которые относятся к этим группам
-
Ответ: Теплокровные: лев, волк, человек, медведь
Холоднокровные: окунь, лягушка, черепаха
Генетический тест «Обмен веществ» | Государственное бюджетное учреждение здравоохранения Детская городская клиническая больница (ГБУЗ ДГКБ г. Краснодара)
Пример заключения
«Генетика обмена веществ»
Разработка способов индивидуализации фитнес-программ по контролю веса, основанных на данных генетического анализа является очень популярным направлением. Некоторые из таких первых подходов уже запатентованы и начинают применяться на практике.
Среди генетических факторов, которые могут повлиять на правильный выбор диеты и интенсивности физических нагрузок, чаще всего анализируют нуклеотидные полиморфизмы генов FABP2, PPARG, ADRB2 и ADRB3. Эти гены, а точнее их генетическая вариабельность, значительно влияют на то, как быстро и эффективно усваиваются питательные вещества, поступающие с пищей, а значит, они могут препятствовать, или помогать достижению и поддержке оптимального веса. Эти генетические вариации (полиморфизмы) не являются патогенными мутациями, они достаточно широко распространены, но, располагая информацией об их наличии или отсутствии, можно точнее оценить индивидуальные особенности своего организма.
Эти гены, а точнее их генетическая вариабельность, значительно влияют на то, как быстро и эффективно усваиваются питательные вещества, поступающие с пищей, а значит, они могут препятствовать, или помогать достижению и поддержке оптимального веса. Эти генетические вариации (полиморфизмы) не являются патогенными мутациями, они достаточно широко распространены, но, располагая информацией об их наличии или отсутствии, можно точнее оценить индивидуальные особенности своего организма.
Так, например, ген FABP2 кодирует белок, связывающий и транспортирующий жирные кислоты в кишечнике. Нуклеотидный вариант («G» или «А») в точке полиморфизма rs1799883 в этом гене приводит к синтезу разных вариантов белка. Один из них лучше связывается с жирными кислотами и, соответственно, люди с таким вариантом гена (генотип GA или AA полиморфизма rs1799883) эффективней усваивают жиры из потребляемой пищи, имеют более высокий индекс массы тела (ИМТ) по сравнению с обладателями альтернативного варианта гена FABP2. Таким пациентам для поддержания оптимального веса рекомендуется питание с пониженным содержанием жиров.
Таким пациентам для поддержания оптимального веса рекомендуется питание с пониженным содержанием жиров.
Ген PPARG, кодирующий гамма-рецептор, активируемый пролифератором пероксисом, отвечает за процессы окисления жирных кислот. Он также влияет на потребность мышечной ткани в глюкозе и ее чувствительность к инсулину. Менее благоприятный вариант полиморфизма rs1801282 (часто называемый как Pro12Ala) приводит к пониженной активности рецептора и, соответственно, к повышению уровня общего холестерина, снижению уровня триглицеридов и повышению чувствительности тканей к инсулину.
Два других популярных гена ADRB2 и ADRB3 кодируют варианты бета-адренергических рецепторов. Встроенные в цитоплазматическую мембрану клетки, они имеют высокую степень сродства к адреналину и регулируют повышение или понижение активности иннервируемой ткани или органа. Активация рецепторов вызывает увеличение интенсивности гликогенолиза в мышцах, увеличение интенсивности секреции инсулина, глюкагона.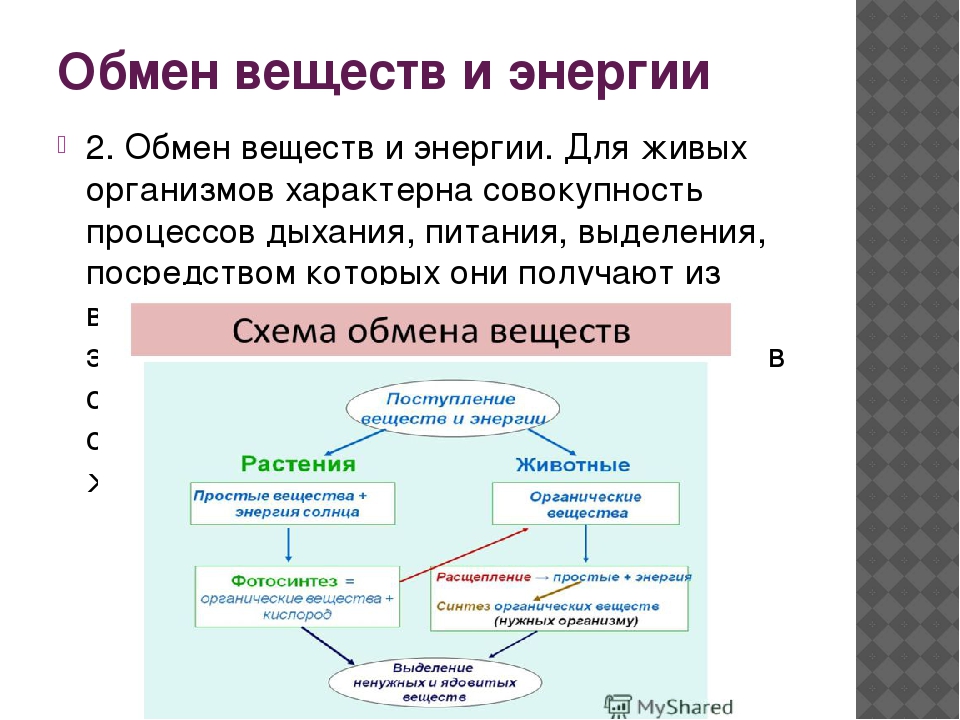 Полиморфизмы rs1042714 и rs4994 в этих генах влияют на эффективность кодируемых ими рецепторов. При менее благоприятных вариантах легче набирается избыточный вес, а снизить его удается только при более интенсивных тренировках.
Полиморфизмы rs1042714 и rs4994 в этих генах влияют на эффективность кодируемых ими рецепторов. При менее благоприятных вариантах легче набирается избыточный вес, а снизить его удается только при более интенсивных тренировках.
Таким образом, информация по вышеуказанным генетическим полиморфизмам может быть весьма ценной для оптимизации усилий по поддержанию веса и других важных показателей в норме. Варианты генов ADRB2 и ADRB3 могут подсказать то, насколько высокоинтенсивными должны быть физические нагрузки для достижения оптимального результата по коррекции веса. А полиморфизмы генов FABP2 и PPARG позволяют оптимизировать профиль питания.
Тест для определения генетических полиморфизмов
«ОБМЕН ВЕЩЕСТВ»
Результаты теста позволяют подобрать индивидуальную диету при снижении веса, а также определить рекомендуемую интенсивность и тип физических нагрузок.
Обмен веществ и превращение энергии в организме человека — основа обеспечения его нормального функционирования — Учебник по Биологии.
 8 класс. Матяш
8 класс. МатяшУчебник по Биологии. 8 класс. Матяш — Новая программа
Тема 1
В чем заключается роль обмена веществ и превращения энергии как основного свойства живого?
Как меняются пищевые и энергетические потребности организма человека в зависимости от вида его деятельности?
Вспомните, какие организмы относятся к гетеротрофам. Какие углеводы запасаются в клетках грибов, растений и животных? Что такое гомеостаз?
Обзор строения и процессов жизнедеятельности человека мы начинаем с обмена веществ и превращения энергии, поскольку эти процессы обеспечивают основу существования нашего организма.
Что такое обмен веществ? Организм человека, как и другие биологические системы, является открытой системой (рис. 31). Вы знаете, что обмен веществ — одно из основных свойств живого. Поэтому обязательным условием существования нашего организма является поступление извне питательных веществ, содержащих в себе энергию (вспомните: организм растений способен улавливать и использовать энергию света).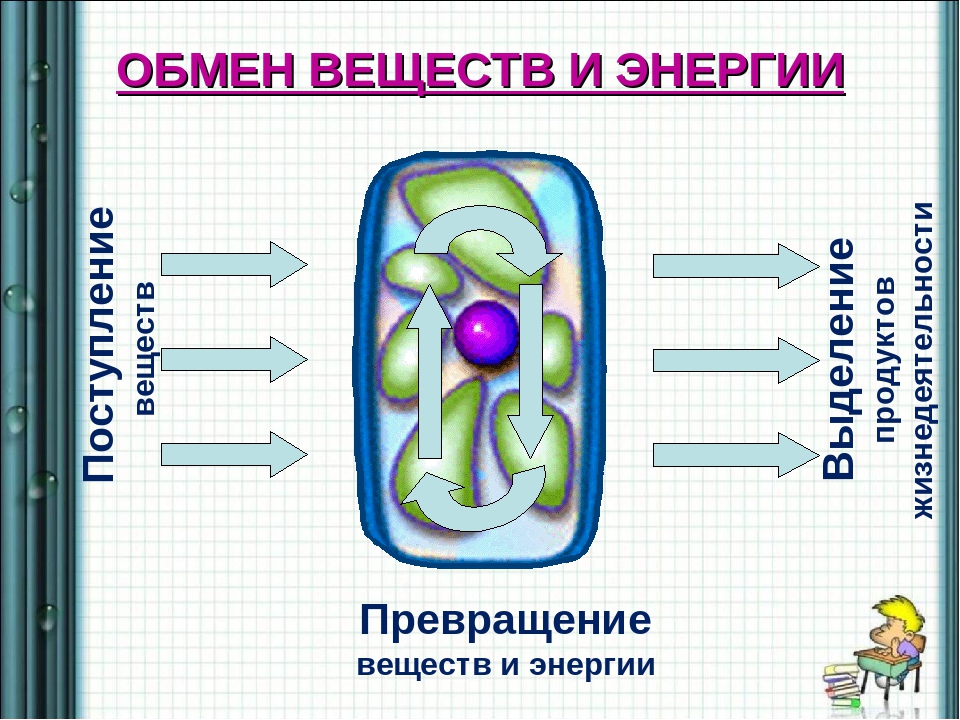 Полученные из внешней среды вещества и энергия подвергаются значительным превращениям в нашем организме. Питательные вещества усваиваются и энергия, накопленная в них, используется. Продукты обмена выводятся наружу (рис. 32).
Полученные из внешней среды вещества и энергия подвергаются значительным превращениям в нашем организме. Питательные вещества усваиваются и энергия, накопленная в них, используется. Продукты обмена выводятся наружу (рис. 32).
Рис. 31. Схема, иллюстрирующая клетку как открытую систему
Рис. 32. Схема обмена веществ в организме человека. Задание. Объясните, какие вещества человек получает из окружающей среды непосредственно, а какие — из продуктов питания; какие вещества организм человека выделяет в окружающую среду
Обмен веществ еще называют метаболизмом. Это основа функционирования любого живого организма. В процессах обмена веществ нашего организма участвуют разные типы соединений — как органические (белки, жиры, углеводы и т. п.), так и неорганические (неорганические кислоты, соли, кислород, углекислый газ, вода и др.). Процессы метаболизма обеспечивают рост и развитие нашего организма, позволяют реагировать на раздражители внешней и внутренней среды, оставлять потомство. Благодаря обмену веществ структуры организма постоянно обновляются, поддерживается постоянство его внутренней среды — гомеостаз.
Благодаря обмену веществ структуры организма постоянно обновляются, поддерживается постоянство его внутренней среды — гомеостаз.
ЗАПОМНИТЕ! Обмен веществ, или метаболизм, — сложная цепь превращений разнообразных соединений в организме, начиная с момента попадания их из внешней среды и заканчивая выведением продуктов распада (рис. 32).
Основу обмена веществ составляют комплексы взаимосвязанных биохимических реакций, которые не могут происходить без участия ферментов.
Что такое ферменты? Ферменты, или энзимы, — это биологически активные вещества, в основном белковой природы, способные влиять на скорость протекания химической реакции. Представьте себе: биохимические реакции при участии ферментов происходят в 106-1012 раз быстрее, чем при их отсутствии. За несколько секунд или даже долей секунды в организме происходит сложная последовательность реакций, каждая из которых требует участия своего специфического фермента. Одни из них ускоряют расщепление сложных органических соединений на более простые, другие — обеспечивают образование собственных веществ организма.
Одни из них ускоряют расщепление сложных органических соединений на более простые, другие — обеспечивают образование собственных веществ организма.
Одной из главных особенностей ферментов является то, что их пространственная структура отвечает пространственной структуре веществ, вступающих в реакцию, подобно тому, как соответствует ключ к замку (рис. 33). Поэтому ферментам присуща специфичность: определенный фермент может обеспечивать один или несколько типов подобных реакций.
Рис. 33. Механизм действия ферментов: 1 — сближение фермента и сложного вещества; 2 — взаимодействие фермента и вещества; 3 — сложное вещество распалось на простые
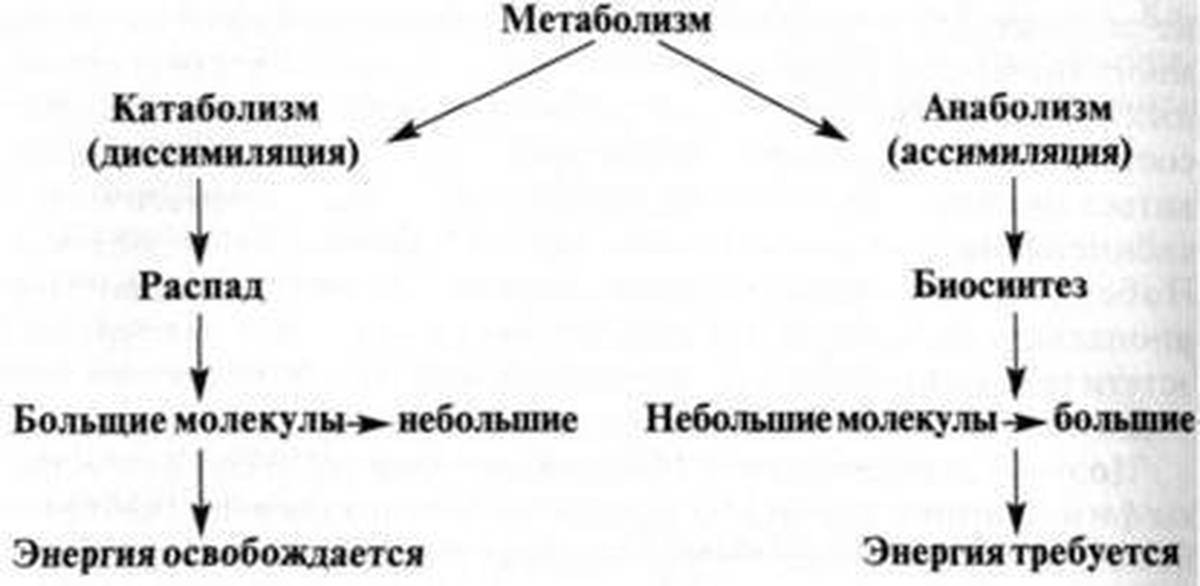
Какое значение для организма имеют питательные вещества? Процессы обмена веществ состоят из реакций двух типов: расщепление сложных органических соединений на более простые — процессы диссимиляции и одновременное образование более сложных соединений из простых — процессы ассимиляции. Обычно эти два процесса в организме человека сбалансированы.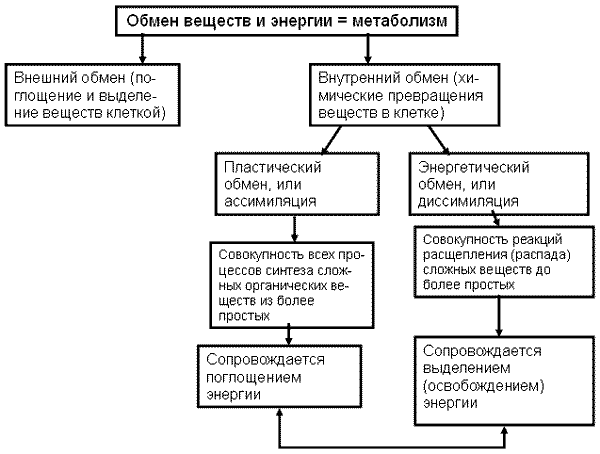 Но в растущем организме процессы ассимиляции должны преобладать над процессами диссимиляции (подумайте почему).
Но в растущем организме процессы ассимиляции должны преобладать над процессами диссимиляции (подумайте почему).
Во время протекания реакций диссимиляции освобождается необходимая для организма энергия. Реакции ассимиляции происходят с расходованием энергии. Поэтому запасы энергии в организме должны постоянно восполняться.
Организм человека способен запасать определенные вещества и соответственно накопленную в них энергию. Некоторое количество жиров запасается в подкожной жировой клетчатке, сальнике и т. п., а углеводов (в виде гликогена) — в клетках печени и мышц. При необходимости эта энергия может быть использована организмом.
Необходимая организму энергия высвобождается в результате окисления или бескислородного расщепления органических соединений (белков, жиров, углеводов). Так, при расщеплении 1 г белков и углеводов выделяется свыше 17 кДж энергии, а 1 г жиров — 38,9 кДж (см. таблицу 1).
Таблица 1
Функциональное значение для организма белков, жиров и углеводов
Питательные вещества | Энергетическая функция в организме человека | Другие функции органических веществ в организме человека | Источник веществ (продукты питания) |
Белки | При окислении 1 г белка высвобождается 17 кДж, или 71,67 ккал энергии | Строительная (клетки и ткани тела человека), регуляторная (гормоны), транспортная (гемоглобин), защитная (антитела, интерферон) | Растительные: бобовые растения (фасоль, бобы, горох, соя). Животные: яйца, икра, молоко, мясо, рыба |
Углеводы | При окислении 1 г углеводов высвобождается 17,6 кДж, или 73,33 ккал энергии | Запасающая (гликоген), строительная (входят в состав гликокаликса) | Растительные: зерновые (хлебо-макаронные изделия), рис, картофель, овощи, фрукты |
Продолжение таблицы 1
Питательные вещества | Энергетическая функция в организме человека | Другие функции органических веществ в организме человека | Источник веществ (продукты питания) |
Жиры | При окислении 1 г жиров высвобождается 38,9 кДж, или 162,08 ккал энергии | Запасающая (жировые депо), теплоизоляционная, защитная (защищают кожу от высыхания и набухания), строительная (входят в состав клеточных мембран) | Растительные масла (подсолнечное, оливковое, кукурузное и т. Животные: масло, сало |
 п.).
п.).Здоровье человека. Для полноценной жизнедеятельности организма человека необходимо энергии приблизительно 10 500 кДж в сутки.
В результате биохимических реакций белки и углеводы могут превратиться в жиры, а жиры — в углеводы. Однако углеводы и жиры никогда не превращаются в белки.
Обмен веществ и превращение энергии в организме человека регулируют нервная и эндокринная (с помощью биологически активных веществ, в основном гормонов) системы. Подробнее о регуляции обмена веществ вы узнаете в § 55.
Ключевые термины и понятия: метаболизм, диссимиляция, ассимиляция, ферменты.
ОБОБЩИМ ЗНАНИЯ
• Одним из условий жизнедеятельности организма человека является обмен веществ и превращение энергии — метаболизм. Он состоит из двух взаимосвязанных процессов: диссимиляции (расщепление сложных органических веществ на более простые с высвобождением энергии) и ассимиляции (образование необходимых организму органических веществ с расходом энергии).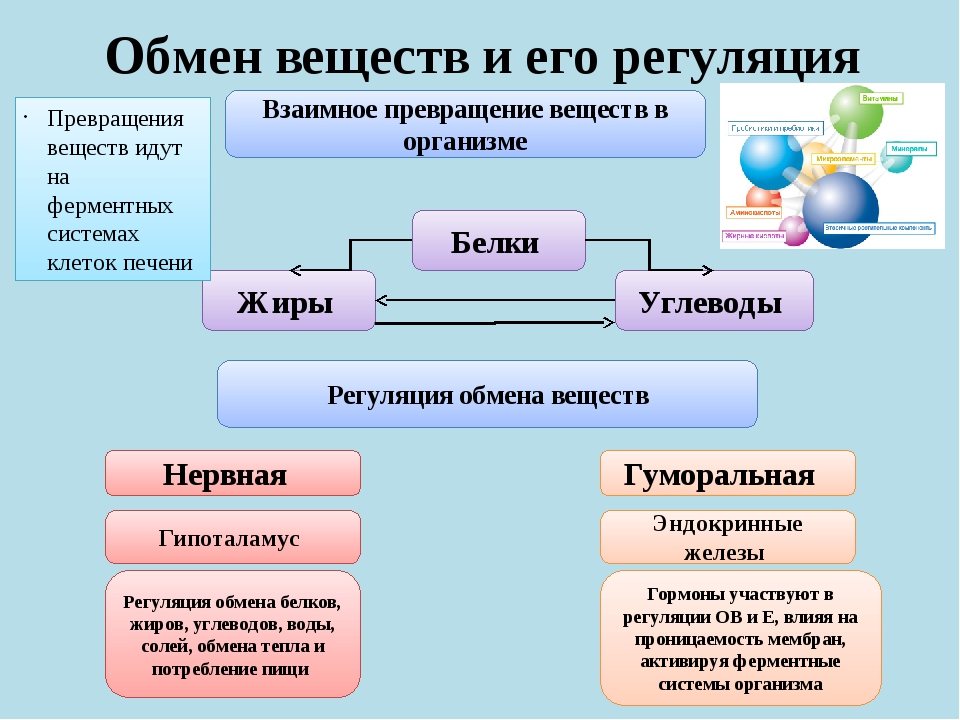
• Ферменты — биологически активные вещества, способные влиять на скорость протекания биохимических реакций.
ПРОВЕРЬТЕ И ПРИМЕНИТЕ ПОЛУЧЕННЫЕ ЗНАНИЯ
Ответьте на вопросы
1. Что такое обмен веществ? 2. Из каких процессов состоит обмен веществ? 3. Для чего нужны запасы энергии в организме человека? 4. Что такое ферменты? Какова их роль в обмене веществ? 5. Что собой представляют процессы диссимиляции? 6. Какова роль процессов ассимиляции в обеспечении метаболизма?
Выберите один правильный ответ
1. Где в организме человека преимущественно может откладываться гликоген: а) в подкожной жировой клетчатке; б) в сальнике; в) в клетках печени; г) в костях?
2. Укажите процессы, с которыми связаны образование и расщепление органических соединений в организме человека: а) фотосинтез и дыхание; б) ассимиляция и диссимиляция; в) дыхание и выделение; г) диссимиляция и дыхание.
Выберите три правильных ответа
Выберите признаки, характерные для ассимиляции.
А превращение веществ | Б энергетические изменения | В место, где происходит процесс |
1 расщепление веществ 2 отсутствие превращений 3 синтез веществ | 1 поглощение энергии 2 высвобождение энергии 3 отсутствие энергетических изменений | 1 клетка 2 межклеточная жидкость 3 полость кишечника |
ПОДУМАЙТЕ. Что общего и отличного в процессах ассимиляции и диссимиляции? Какая связь существует между ними?
С помощью взрослых выполните исследовательский практикум.
Самонаблюдение за соотношением массы тела и роста
Оборудование и материалы: напольные весы, сантиметровая лента.
1. Станьте ровно у стены без обуви, касаясь ее тремя точками: на уровне пяток, ягодичных мышц и лопаток. Голову держите прямо. С помощью сантиметровой ленты измерьте свой рост с точностью до 0,5 см. Данные запишите в таблицу.
Голову держите прямо. С помощью сантиметровой ленты измерьте свой рост с точностью до 0,5 см. Данные запишите в таблицу.
2. Взвесьтесь на напольных весах. Данные занесите в таблицу.
3. Определите свой массо-ростовой индекс. Для этого показатель массы тела (г) разделите на показатель роста. Каждому сантиметру роста должно соответствовать 350-400 г массы у мальчиков и 375-425 г у девочек. Если показатели меньше, то можно говорить о недостаточной массе, если больше — об избыточной. Проанализируйте, почему увеличилась масса тела: из-за жировых отложений или в результате развития мышц.
4. Определите свой росто-массовый показатель (кг), вычитая из показателя роста цифру 100, если рост равен 155-164 см, или цифру 110, если рост равняется 165-185 см.
Процессы обмена веществ | Параграф 37
«Биология. Человек. 9 класс». А.С. Батуев и др.
Вопрос 1.
Процесс образования органических веществ и кислорода из неорганических — углекислого газа и воды в хлоропластах на свету называется фотосинтезом.
Вопрос 2.
Процессы обмена веществ происходят в клетках: распад органических веществ, сопровождающийся выделением энергии, — в митохондриях; образование органических веществ, например, белков, — на рибосомах.
Вопрос 3.
Главное отличие живого организма от неживого — наличие у первого обмена веществ.
Вопрос 4.
Пластический обмен (ассимиляция) — совокупность процессов биосинтеза, когда из простых веществ синтезируются более сложные и происходит накопление энергии химической связи.
Энергетический обмен (диссимиляция) — совокупность ферментативных процессов расщепления сложных органических веществ в организме, сопровождающихся выделением энергии.
Вопрос. 5.
Пластический обмен преобладает в период роста и развития организма.
Вопрос 6.
Уровень обмена веществ в организме зависит от:
а) состояния здоровья;
б)возраста;
в) эмоционального состояния;
г) степени усталости;
д) физической нагрузки.
Вопрос 7.
Обмен веществ. Регуляция. (см. схему 13.).
Рис. 13. Регуляция обмена веществ
Изменение концентрации гормона в крови —> возбуждение нейронов гипоталамуса —> регуляция функции гипофиза —> нормализация деятельности железы —> регуляция уровня обмена веществ.
1 (!)
где
E — рост и развитие, процессы жизнедеятельности (дыхание, сердцебиение), движения, теплообразование (поддержание постоянной температуры тела), обмен веществ.
2 (!). В состоянии покоя потребности организма в энергии незначительны. Расход энергии возрастает при работе. Это объясняется тем, что для мышечных сокращений или проведения нервных импульсов необходима энергия, которая образуется в клетках при окислении органических веществ. Чем больше физическая нагрузка, тем больше энергии необходимо мышцам, тем интенсивнее идут процессы обмена веществ в организме (учащаются сердцебиение и дыхание).
План § 37
Две стороны, единого процесса обмена веществ.
1. Единство живой и неживой природы.
а) Образование органических веществ из неорганических (растения — фотосинтез).
б) Использование органических веществ и их распад.
2. Обмен веществ — основная функция живого организма.
а) Определение понятия «обмен веществ».
б) Распад органических веществ с выделением энергии.
в) Расход энергии.
г) Пища — источник органических веществ.
д) Главное отличие живого от неживого.
3. Пластический обмен.
4. Энергетический обмен.
5. Единство двух противоположных процессов — пластического и энергетического обменов.
6. Регуляция обмена веществ.
а) Нервная (центры в промежуточном мозге).
б) Гуморальная (влияние гормонов).
7. Потребность организма в энергии.
Зависимость потребности от вида деятельности; возраста.
Что такое метаболический путь? — Определение и пример — Видео и стенограмма урока
Существует два основных типа метаболических путей: катаболический и анаболический. Катаболические пути выделяют энергию, расщепляя молекулы на более простые молекулы. Клеточное дыхание — один из примеров катаболического пути. Во время клеточного дыхания сахар поглощается клеткой и расщепляется, чтобы высвободить энергию, которая позволяет нам жить.
Другие типы катаболических путей включают лимонный цикл или цикл Кребса, где ацетат макроэлементов, таких как молекулы белков, жиров и углеводов, подвергается окислению.Конечным результатом является углекислый газ химического соединения. Гликолиз — это еще один тип катаболического пути, при котором организмы и растения накапливают и высвобождают глюкозу и другую энергию сахара для образования высокоэнергетической молекулы, известной как аденозинтрифосфат (АТФ). Биологи называют АТФ «энергетической валютой жизни», потому что он накапливает энергию, необходимую нам для повседневной работы. Процесс гликолиза используется для выработки энергии по катаболическому пути.
Процесс гликолиза используется для выработки энергии по катаболическому пути.
В то время как ферменты катаболических путей расщепляют молекулы и высвобождают энергию, ферменты анаболических путей или биосинтетических реакций нуждаются в энергии для изменения или преобразования молекул в более сложные молекулы или макромолекулы.Например, аминокислоты можно использовать для создания белков, углекислый газ можно использовать для производства сахара, а нуклеиновые кислоты можно использовать для создания новых цепей ДНК, которые можно найти почти в каждой из ваших клеток.
Возможно, вы слышали термин «анаболический» в более негативном значении по отношению к спорту или культуристам. Некоторые спортсмены или бодибилдеры принимают анаболические стероиды для создания более крупных и сильных мышц. Хотя использование анаболических стероидов связано с риском для здоровья и несправедливыми конкурентными преимуществами, оно служит примером того, как брать что-то меньшее и делать это больше и сложнее.
Следующий белок был создан анаболическим путем:
Все живые организмы имеют определенные метаболические пути, которые используются для расщепления или создания молекул. Без них мы не были бы живы. Некоторые из этих путей и процессов очень сложны и выходят за рамки цели и объема этого урока. Подумайте о них как о повседневных химических процессах и реакциях, происходящих в нашем теле, которые позволяют нам дышать, есть, двигаться и думать.
Резюме урока
Давайте рассмотрим:
- Метаболизм — это сумма химических реакций, происходящих в нашем организме.
- Метаболические пути — это химические реакции, которые имеют место для создания и использования энергии.
- Ферменты в химических реакциях обладают способностью разрушать, накапливать или останавливать химические реакции.
- Катаболические пути включают расщепление молекул с высвобождением энергии (например,г. , посредством клеточного дыхания).
- Анаболические пути включают создание молекул для создания более сложных молекул (например, путем создания белков).
 , посредством клеточного дыхания).
, посредством клеточного дыхания).| Катаболический | Анаболический |
|---|---|
| * Высвобождение энергии за счет расщепления молекул на более простые формы * Включает цикл Кребса и гликолиз | * Биосинтетические реакции * Создание молекул для создания более сложных молекул * Примеры: аминокислоты создаются для производства белков, углекислый газ создается для производства сахара |
Результаты обучения
По завершении этого урока вы должны уметь:
- Объяснять, что такое метаболический путь и его значение для жизни
- Проведите различие между катаболическими и анаболическими путями метаболизма и приведите примеры каждого из них.
границ | Определение метаболического пути генов растений на основе филогенетического профилирования — технико-экономическое обоснование
Введение
Развитие понимания метаболизма растений — центральная цель исследований растений.Чем лучше мы сможем оценить метаболические возможности растений и то, как они регулируют свою метаболическую активность, тем лучше мы сможем использовать множество продуктов, а также защитить их хрупкие экосистемы. В принципе, должна быть возможность оценить метаболическую способность растения на основе знания всех возможных метаболических реакций, которые, в свою очередь, кодируются репертуаром генов ферментов в соответствующем геноме. Таким образом, полная и точная аннотация генома имеет первостепенное значение для всестороннего понимания метаболизма растений.Однако надежная аннотация функционального гена не является тривиальной задачей, и наши текущие знания о возможных метаболических путях неполны. Мы пока не можем просто проверить наличие «путей из учебников» с помощью точной аннотации генов. Новые пути, в частности, в контексте путей вторичных метаболитов, все еще открываются, что, однако, требует значительных экспериментальных усилий, как продемонстрировано в открытии пути стриголактона у растений (Alder et al., 2012).
Новые пути, в частности, в контексте путей вторичных метаболитов, все еще открываются, что, однако, требует значительных экспериментальных усилий, как продемонстрировано в открытии пути стриголактона у растений (Alder et al., 2012).
Учитывая высокую стоимость и огромные усилия по экспериментальной аннотации функций генов, вычислительная сравнительная геномика остается основной стратегией приписывания функций генам в растениях.Установленные методы функционального назначения направлены на биоинформатическое прогнозирование функций белков еще не аннотированных видов путем поиска белков, сходных с последовательностями, несущих надежно аннотированные, в идеале экспериментально подтвержденные функции. Предполагая, что высокое сходство последовательностей обеспечивает сходство функций, функциональные аннотации переносятся с охарактеризованного гена на новый, еще не охарактеризованный ген (Lohse et al., 2014).
Однако гены, активные в одном и том же биохимическом пути, обычно будут выполнять различные ферментативные функции и, таким образом, обычно не будут демонстрировать никакого сходства аминокислотных последовательностей друг с другом, хотя сообщается о слабой, но заметной корреляции между метаболическим путем и расстояниями между последовательностями ферментов и белков. к постепенному расширению метаболизма (Schutte et al., 2010). Несмотря на это наблюдение, методы, основанные на гомологии последовательностей, обычно имеют ограниченное применение для создания связей между компонентами, которые выполняют разные функции как часть одного и того же пути. Способы функционального прогнозирования, основанные на гомологии, также по своей сути ограничены набором известных функциональных аннотаций.
к постепенному расширению метаболизма (Schutte et al., 2010). Несмотря на это наблюдение, методы, основанные на гомологии последовательностей, обычно имеют ограниченное применение для создания связей между компонентами, которые выполняют разные функции как часть одного и того же пути. Способы функционального прогнозирования, основанные на гомологии, также по своей сути ограничены набором известных функциональных аннотаций.
Было разработано несколько стратегий для установления функциональных связей между генами, выполняющими разные функции. Они полагаются на наблюдаемую физическую близость генов, связанных с путями, происходящих из структур генома бактериального оперона (Osbourn, 2010; Chu et al., 2011) или выраженная коэкспрессия генов (Gachon et al., 2005; Wisecaver et al., 2016). Полногеномные исследования ассоциации позволяют идентифицировать гены, обычно участвующие или регулирующие биосинтез определенного метаболита (Yencho et al., 1998; Schauer et al., 2006; Kliebenstein, 2009).
Филогенетическое профилирование предлагает еще один подход к обнаружению функциональных ассоциаций генов. Филогенетическое профилирование было разработано на основе представления о том, что гены, участвующие в одном и том же метаболическом пути или обычно участвующие в одном и том же функциональном процессе, вероятно, будут эволюционировать коррелированным образом (Gaasterland and Ragan, 1998; Pellegrini et al., 1999). Для данного процесса все его существенные элементы (гены) либо присутствуют — поскольку все они необходимы для выполнения определенной функции — либо все отсутствуют, потому что, если какой-либо компонент отсутствует, все остальные компоненты больше не могут функционировать, поднимая эволюционный давление на них должно быть сохранено. Концепция филогенетического профилирования была впервые протестирована для прогнозирования функциональных взаимосвязей между белками E. coli на основе их филогенетического профиля в 16 полностью секвенированных организмах, включая S. cerevisiae, B. subtilis и H. influenza (Pellegrini et al., 1999). Помимо объединения функционально разнообразных генов в общие процессы, филогенетическое профилирование также предлагает путь к предоставлению аннотации для не охарактеризованных иначе последовательностей. Даже без каких-либо знаний о функции конкретного гена, знание того, что он функционально связан с другими генами, уже дает ценную информацию и точки входа для дальнейшей функциональной характеристики.
cerevisiae, B. subtilis и H. influenza (Pellegrini et al., 1999). Помимо объединения функционально разнообразных генов в общие процессы, филогенетическое профилирование также предлагает путь к предоставлению аннотации для не охарактеризованных иначе последовательностей. Даже без каких-либо знаний о функции конкретного гена, знание того, что он функционально связан с другими генами, уже дает ценную информацию и точки входа для дальнейшей функциональной характеристики.
После новаторской работы Пеллегрини и его сотрудников основная концепция филогенетического профилирования нашла множество применений, например.g., чтобы предсказать белок-белковые взаимодействия (Pagel et al., 2004; Kim and Subramaniam, 2006) или идентифицировать специфические ферменты, участвующие в биосинтезе определенных метаболитов у грибов (Ternes et al., 2006). С момента своего создания методологическая основа филогенетического профилирования была уточнена путем тестирования пригодности взаимоотношений ортологичных и паралоговых генов (Skunca et al. , 2013), путем использования структурной информации для улучшенного определения гомологии (Ranea et al., 2007), путем внедрения новых метрик расстояния для измерения сходства профилей (Vert, 2002), а также путем оценки необходимого количества видов для успешных приложений филогенетического профилирования (Škunca and Dessimoz, 2015).Недавно был разработан ProtPhylo, удобный веб-сервис для поиска белков, которые, возможно, связаны с эталонным белком согласно филогенетическому профилированию (Cheng and Perocchi, 2015). ProtPhylo также позволяет устанавливать связи между видоспецифическими фенотипами и ассоциированными белками-кандидатами.
, 2013), путем использования структурной информации для улучшенного определения гомологии (Ranea et al., 2007), путем внедрения новых метрик расстояния для измерения сходства профилей (Vert, 2002), а также путем оценки необходимого количества видов для успешных приложений филогенетического профилирования (Škunca and Dessimoz, 2015).Недавно был разработан ProtPhylo, удобный веб-сервис для поиска белков, которые, возможно, связаны с эталонным белком согласно филогенетическому профилированию (Cheng and Perocchi, 2015). ProtPhylo также позволяет устанавливать связи между видоспецифическими фенотипами и ассоциированными белками-кандидатами.
В этом исследовании мы проверили применимость филогенетического профилирования конкретно к проблеме назначения метаболических путей растений. Мы были особенно заинтересованы в функциональном назначении генов вторичного метаболизма, специфичных для растений, и в оценке точности филогенетического профилирования с учетом имеющихся в настоящее время последовательностей генома растений и информации об аннотациях. Вторичные метаболиты растений представляют особый экономический и медицинский интерес, поскольку многие из них обладают полезными свойствами для использования в питании и в медицине (Singh and Bhat, 2003; Schmidt et al., 2007). Эффективная и надежная оценка запасов вторичных метаболитов в растении имеет огромный экономический потенциал и обеспечивает основу для разработки целевых путей (Verpoorte и Memelink, 2002; Oksman-Caldentey and Inze, 2004). В отличие от путей первичного метаболизма, пути вторичного метаболизма часто функционируют как независимые единицы с низкими уровнями функциональных зависимостей и, что более важно для обоснования филогенетического профилирования, влияют на другие биохимические функции (Hartmann, 1996; Higashi and Saito, 2013).Таким образом, набор генов, связанных с конкретным вторичным путем, может появляться и исчезать независимо от других путей. А поскольку специфические метаболические пути происходят только у подгруппы видов (Pichersky and Gang, 2000), подход филогенетического профилирования должен идеально подходить для идентификации вторичных метаболических путей на основе филогенетического профиля присутствия-отсутствия их ферментов.
Вторичные метаболиты растений представляют особый экономический и медицинский интерес, поскольку многие из них обладают полезными свойствами для использования в питании и в медицине (Singh and Bhat, 2003; Schmidt et al., 2007). Эффективная и надежная оценка запасов вторичных метаболитов в растении имеет огромный экономический потенциал и обеспечивает основу для разработки целевых путей (Verpoorte и Memelink, 2002; Oksman-Caldentey and Inze, 2004). В отличие от путей первичного метаболизма, пути вторичного метаболизма часто функционируют как независимые единицы с низкими уровнями функциональных зависимостей и, что более важно для обоснования филогенетического профилирования, влияют на другие биохимические функции (Hartmann, 1996; Higashi and Saito, 2013).Таким образом, набор генов, связанных с конкретным вторичным путем, может появляться и исчезать независимо от других путей. А поскольку специфические метаболические пути происходят только у подгруппы видов (Pichersky and Gang, 2000), подход филогенетического профилирования должен идеально подходить для идентификации вторичных метаболических путей на основе филогенетического профиля присутствия-отсутствия их ферментов. Кроме того, с огромным увеличением доступных данных по полному геному, теперь может быть доступна необходимая база данных для проверки и практического применения филогенетического профилирования.
Кроме того, с огромным увеличением доступных данных по полному геному, теперь может быть доступна необходимая база данных для проверки и практического применения филогенетического профилирования.
Мы внедрили комплексную и тщательную схему тестирования, охватывающую 39 видов растений и 40 960 функционально охарактеризованных генов ферментов. Доказав успешность при тестировании на конкретных путях, демонстрация общей пригодности филогенетического профилирования в настоящее время серьезно затруднена из-за нехватки вторичных путей, встречающихся только в подмножестве видов. Аннотировано, что многие пути происходят почти у всех видов растений, и, следовательно, сама основа филогенетического профилирования — присутствие только в подмножестве видов — часто не выполняется, что демонстрирует истинный потенциал проблем филогенетического профилирования.Кроме того, мы демонстрируем, что наше разделение путей на изолированные единицы критически влияет на применимость филогенетического профилирования. Тем не менее, мы считаем, что это исследование представляет собой ценный систематический тест на осуществимость, подчеркивающий необходимость продолжения экспериментальной работы по аннотации, и в то же время демонстрирующий, что филогенетический профиль открывает огромные перспективы для заполнения пробелов в наших знаниях о метаболизме растений.
Тем не менее, мы считаем, что это исследование представляет собой ценный систематический тест на осуществимость, подчеркивающий необходимость продолжения экспериментальной работы по аннотации, и в то же время демонстрирующий, что филогенетический профиль открывает огромные перспективы для заполнения пробелов в наших знаниях о метаболизме растений.
Материалы и методы
Филогенетическое профилирование работает путем присвоения определенной кодируемой геном молекулярной (здесь ферментативной) функции, присутствующей или отсутствующей у данного вида.Затем предполагается, что гены со сходными профилями присутствия-отсутствия у нескольких видов участвуют в одном и том же функциональном процессе, в нашем случае метаболическом пути. Вызов присутствия / отсутствия основан на представлении о том, что гены, сходные с последовательностями, выполняют ту же функцию. Следовательно, необходимо определить порог приемлемого уровня сходства последовательностей для предположения идентичной функции. Кластеризация всех генов, кодируемых в данном наборе видов на основе взаимосвязи их гомологии последовательностей, приводит к получению наборов генов с предполагаемой идентичной функцией.Принадлежность к видам каждого члена кластера будет определять филогенетический профиль данного кластера. Здесь мы называем такие кластеры семействами генов или, в более общем смысле, объектами генов. Семейства / объекты генов также могут состоять из одного члена гена, который будет обозначаться как одиночные. Следуя логике филогенетического профилирования, семейства генов должны (i) кодировать одну и только метаболическую функцию, (ii) разные семейства генов кодировать разные функции и (iii) семейства генов с идентичным филогенетическим профилем должны участвовать в одной и той же метаболической функции.
Кластеризация всех генов, кодируемых в данном наборе видов на основе взаимосвязи их гомологии последовательностей, приводит к получению наборов генов с предполагаемой идентичной функцией.Принадлежность к видам каждого члена кластера будет определять филогенетический профиль данного кластера. Здесь мы называем такие кластеры семействами генов или, в более общем смысле, объектами генов. Семейства / объекты генов также могут состоять из одного члена гена, который будет обозначаться как одиночные. Следуя логике филогенетического профилирования, семейства генов должны (i) кодировать одну и только метаболическую функцию, (ii) разные семейства генов кодировать разные функции и (iii) семейства генов с идентичным филогенетическим профилем должны участвовать в одной и той же метаболической функции.
Следуя этой логике, были реализованы следующие этапы обработки и подходы к проверке ее достоверности. (A) На основе информации, доступной в базе данных Ensembl Plants (Kersey et al., 2016), и дополнительных этапов фильтрации, были созданы семейства генов для полного известного реестра генов 39 видов растений. (B) Для каждого семейства генов были созданы филогенетические профили на основе видового происхождения всех его последовательностей. (C) Генные семейства были протестированы на предмет правильного отражения общей и уникальной функции, а также того, предполагают ли идентичные филогенетические профили различных семейств генов участие в общем процессе; я.е. метаболический путь. Тестирование производительности филогенетического профилирования как средства определения ассоциаций путей было основано на данных аннотаций, доступных в базах данных Ensembl Plants (Kersey et al., 2016), а также KEGG (Kanehisa and Goto, 2000). (D) Наблюдаемые результаты производительности сравнивали с рандомизированными данными для оценки статистической значимости.
(B) Для каждого семейства генов были созданы филогенетические профили на основе видового происхождения всех его последовательностей. (C) Генные семейства были протестированы на предмет правильного отражения общей и уникальной функции, а также того, предполагают ли идентичные филогенетические профили различных семейств генов участие в общем процессе; я.е. метаболический путь. Тестирование производительности филогенетического профилирования как средства определения ассоциаций путей было основано на данных аннотаций, доступных в базах данных Ensembl Plants (Kersey et al., 2016), а также KEGG (Kanehisa and Goto, 2000). (D) Наблюдаемые результаты производительности сравнивали с рандомизированными данными для оценки статистической значимости.
Все 39 видов растений ансамбля, рассмотренных в этом исследовании, вместе с аннотацией о присутствии в них KEGG, перечислены в Таблице 1.
Таблица 1. видов растений и геномов, использованных в этом исследовании.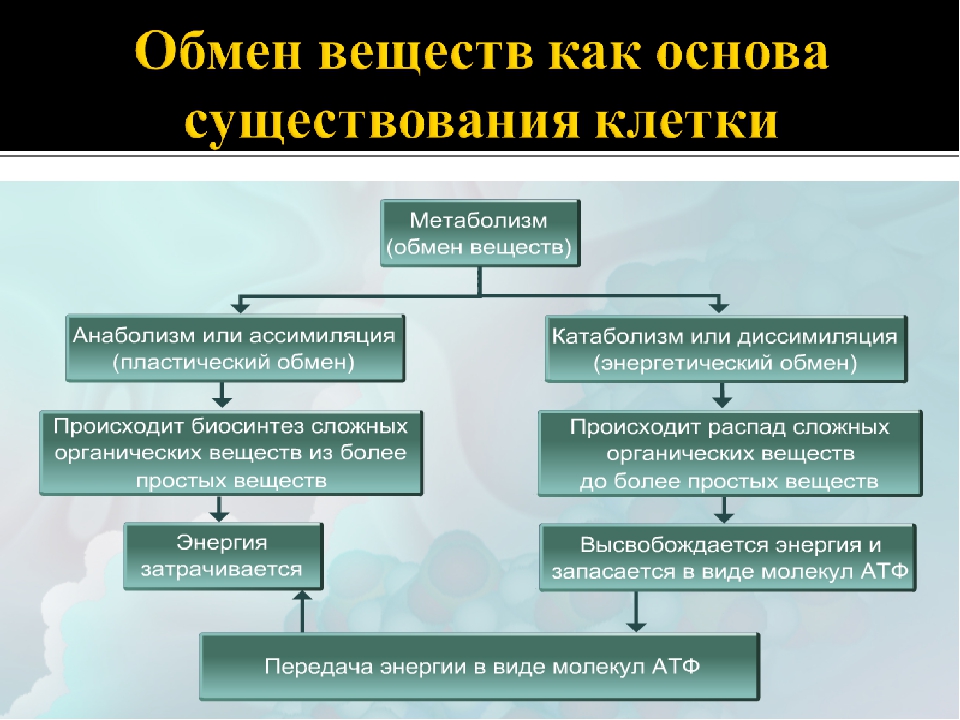
Информация о последовательности и гомологии
Поскольку нас интересовала ферментативная активность, все гены, их последовательности, функции и меры их попарного сходства рассматривались на основе их соответствующих белковых последовательностей. Информация о гомологии и белковые последовательности ферментов метаболического пути для 39 видов растений (таблица 1), доступная в базе данных Plant Mart, была загружена с Ensembl Plants (Kersey et al., 2016).Инструмент Biomart использовался для отбора видов и извлечения всех паралогичных и ортологичных генов, их идентичности последовательностей, определения достоверности гомологии и их номеров EC (Kinsella et al., 2011).
Кластеризация генов в генные семейства
Все гены были сгруппированы в соответствии с их гомологией на основе последовательностей для определения генов и выполняемых ими функций как присутствующих или отсутствующих у данного вида. Набор взаимно гомологичных последовательностей образует набор генов, который, по сути, можно рассматривать как единый объект, состоящий либо из нескольких последовательностей, называемых семейством генов, либо из одной последовательности в случаях, когда гомологичная последовательность не была обнаружена. Последние упоминаются как одиночные. Каждому такому объекту (семейству генов или отдельному объекту) будет затем присвоен филогенетический профиль, отражающий его присутствие или отсутствие во всех рассматриваемых видах растений вместе с соответственно выполняемой ферментативной функцией. Мы действуем исходя из предположения, что все последовательности, сгруппированные вместе в семейство генов, выполняют одну и ту же ферментативную функцию.
Последние упоминаются как одиночные. Каждому такому объекту (семейству генов или отдельному объекту) будет затем присвоен филогенетический профиль, отражающий его присутствие или отсутствие во всех рассматриваемых видах растений вместе с соответственно выполняемой ферментативной функцией. Мы действуем исходя из предположения, что все последовательности, сгруппированные вместе в семейство генов, выполняют одну и ту же ферментативную функцию.
База данных Ensembl Plants предоставляет список ортологичных и паралогичных генов, а также значения парной идентичности последовательностей для всех включенных генов.Кроме того, предоставляется значение достоверности ортологии (низкое или высокое), полученное при сравнении с филогенетическим деревом (Vilella et al., 2009). В этом исследовании использовались только ортологические отношения с высокой степенью достоверности.
Парные отношения гомологии между всеми генами всех 39 видов были отфильтрованы с учетом процента идентичности последовательностей относительно более короткой из двух сравниваемых последовательностей, как указано в Ensembl, устанавливающем два разных порога идентичности последовательностей 30% и 70% (выравнивание белков), соответственно. Все гены, не принадлежащие к какому-либо множественному семейству генов, считались генами-одиночками. Комбинируя как отношение гомологии, о котором сообщают Ensembl, так и идентичность последовательностей, была создана сеть с генами, представляющими ее узлы, которые связаны, если они являются гомологичными и проходят установленный порог идентичности последовательностей. Связанные компоненты этой генной сети, обнаруженные с помощью пакета R igraph (Csardi and Nepusz, 2006), считаются семействами генов. Компонент связности определяется как подграф, в котором все узлы связаны, т.е.е. существует путь между всеми узлами подграфа. В описанной выше процедуре паралогичные и ортологичные отношения генов рассматривались одинаково. Две кластеры генов в генные семейства на основе сетей идентичности последовательностей 30% или 70% будут называться Network30 и Network70 соответственно.
Все гены, не принадлежащие к какому-либо множественному семейству генов, считались генами-одиночками. Комбинируя как отношение гомологии, о котором сообщают Ensembl, так и идентичность последовательностей, была создана сеть с генами, представляющими ее узлы, которые связаны, если они являются гомологичными и проходят установленный порог идентичности последовательностей. Связанные компоненты этой генной сети, обнаруженные с помощью пакета R igraph (Csardi and Nepusz, 2006), считаются семействами генов. Компонент связности определяется как подграф, в котором все узлы связаны, т.е.е. существует путь между всеми узлами подграфа. В описанной выше процедуре паралогичные и ортологичные отношения генов рассматривались одинаково. Две кластеры генов в генные семейства на основе сетей идентичности последовательностей 30% или 70% будут называться Network30 и Network70 соответственно.
Филогенетические профили
Филогенетических профилей были созданы для каждого генного объекта, включая семейства генов и ген-одиночку. Генные объекты, кодирующие определенную функцию, считались присутствующими у определенного вида, если в нем был обнаружен хотя бы один из его генов-членов, в противном случае генный объект считался отсутствующим.Затем вызов присутствия / отсутствия для всех 39 рассматриваемых видов растений представляет филогенетический профиль генного объекта, кодируемый единицами (указывающими на присутствие) и нулями (указывающими на отсутствие). Затем генные объекты с идентичными филогенетическими профилями были сгруппированы вместе. Поскольку мы предположили, что каждый генный объект связан с одной уникальной ферментативной функцией, тогда предполагается, что генные объекты, сгруппированные вместе по идентичным филогенетическим профилям, участвуют в одном и том же ферментативном процессе (пути). Достоверность этого утверждения находится в центре внимания данного исследования и проверяется с помощью следующей статистической процедуры.
Генные объекты, кодирующие определенную функцию, считались присутствующими у определенного вида, если в нем был обнаружен хотя бы один из его генов-членов, в противном случае генный объект считался отсутствующим.Затем вызов присутствия / отсутствия для всех 39 рассматриваемых видов растений представляет филогенетический профиль генного объекта, кодируемый единицами (указывающими на присутствие) и нулями (указывающими на отсутствие). Затем генные объекты с идентичными филогенетическими профилями были сгруппированы вместе. Поскольку мы предположили, что каждый генный объект связан с одной уникальной ферментативной функцией, тогда предполагается, что генные объекты, сгруппированные вместе по идентичным филогенетическим профилям, участвуют в одном и том же ферментативном процессе (пути). Достоверность этого утверждения находится в центре внимания данного исследования и проверяется с помощью следующей статистической процедуры.
Информация о ферментативном пути
Данные метаболического пути и функциональной аннотации для всех видов, общих для KEGG и Ensembl Plants, были загружены из базы данных KEGG (Kanehisa and Goto, 2000). Функциональная аннотация, полученная из базы данных KEGG, была отнесена ко всем генам и их соответственно кодируемым белкам. Для всех путей вторичных и первичных метаболитов растений, содержащихся в KEGG, приведены номера карт путей, относящиеся к фактическим биохимическим путям. В общей сложности 40 960 генов метаболических ферментов от 24 видов растений с доступным EC-номером от Ensembl Plants были доступны для анализа пути.
Функциональная аннотация, полученная из базы данных KEGG, была отнесена ко всем генам и их соответственно кодируемым белкам. Для всех путей вторичных и первичных метаболитов растений, содержащихся в KEGG, приведены номера карт путей, относящиеся к фактическим биохимическим путям. В общей сложности 40 960 генов метаболических ферментов от 24 видов растений с доступным EC-номером от Ensembl Plants были доступны для анализа пути.
Были протестированы два уровня назначения метаболических путей генов: метаболические классы и метаболические пути. Метаболические классы были взяты в соответствии с базой данных KEGG и включают 10 классов первичных и вторичных метаболических путей: метаболизм аминокислот, биосинтез других вторичных метаболитов, углеводный обмен, энергетический обмен, биосинтез и метаболизм гликанов, метаболизм липидов, метаболизм кофакторов и витаминов, метаболизм других аминокислот, метаболизм терпеноидов и поликетидов и метаболизм нуклеотидов.Была использована более подробная классификация генов ферментов путем рассмотрения 94 фактических карт путей, связанных с 10 классами путей, доступных от KEGG, которые классифицируются как «метаболизм» и имеют ненулевое количество назначенных генов растений (см. Дополнительную таблицу 1) . Гены учитывались в отношении путей вторичного метаболизма только в том случае, если было аннотировано, что они участвуют только во вторичных путях метаболизма. Гены, аннотированные как для первичных, так и для вторичных путей метаболизма, считались генами первичного метаболизма.Класс путей «Обзор» и связанные с ним четыре подробные карты путей не рассматривались, поскольку они могут считаться неспецифическими и также не содержались в данных по растениям Ensembl. Пути, отнесенные к классам «метаболизм терпеноидов и поликетидов» или «биосинтез других вторичных метаболитов», рассматривались как пути вторичного метаболизма. В общей сложности 31 карта KEGG, 17 из которых несли аннотации генов растений, рассматривались как вторичные, а все остальные — как первичные метаболические пути.
Дополнительную таблицу 1) . Гены учитывались в отношении путей вторичного метаболизма только в том случае, если было аннотировано, что они участвуют только во вторичных путях метаболизма. Гены, аннотированные как для первичных, так и для вторичных путей метаболизма, считались генами первичного метаболизма.Класс путей «Обзор» и связанные с ним четыре подробные карты путей не рассматривались, поскольку они могут считаться неспецифическими и также не содержались в данных по растениям Ensembl. Пути, отнесенные к классам «метаболизм терпеноидов и поликетидов» или «биосинтез других вторичных метаболитов», рассматривались как пути вторичного метаболизма. В общей сложности 31 карта KEGG, 17 из которых несли аннотации генов растений, рассматривались как вторичные, а все остальные — как первичные метаболические пути.
Оценка принадлежности генного семейства
Скорректированный индекс Рэнда (Hubert and Arabie, 1985) применяли для оценки достоверности присвоения семейств генов по сравнению с известными метаболическими функциями генов. Функция adjustRandIndex пакета R mclust (Fraley and Raftery, 1999) была использована для сравнения кластеризации генов в семейства генов, предполагая идентичную функцию с этими кластерами на основе аннотации номера EC, отражающей истинную функцию. Результирующий индекс Rand оценивает степень соответствия обеих классификаций, где ноль означает случайность, а значение единицы означает полное совпадение. Множественные / неоднозначные аннотации номеров EC обрабатывались как отдельные истинные функциональные аннотации, так что только тогда считалось, что два гена выполняют одну и ту же функцию, если оба имеют одинаковый набор номеров EC.
Функция adjustRandIndex пакета R mclust (Fraley and Raftery, 1999) была использована для сравнения кластеризации генов в семейства генов, предполагая идентичную функцию с этими кластерами на основе аннотации номера EC, отражающей истинную функцию. Результирующий индекс Rand оценивает степень соответствия обеих классификаций, где ноль означает случайность, а значение единицы означает полное совпадение. Множественные / неоднозначные аннотации номеров EC обрабатывались как отдельные истинные функциональные аннотации, так что только тогда считалось, что два гена выполняют одну и ту же функцию, если оба имеют одинаковый набор номеров EC.
Статистика сравнения филогенетических профилей
Тест на обогащение идентичных профилей в пределах разных путей
Для каждого из 10 метаболических классов и 94 метаболических путей были получены соответственно аннотированные гены ферментов. Обратите внимание, что гены могут происходить от любого из 24 видов растений, аннотированных в KEGG, и при этом относиться к одному и тому же метаболическому классу или пути. Для каждого результирующего набора из n и ферментных генов, связанных с одним конкретным метаболическим классом или путем, был определен связанный набор из n f генных объектов (семейства генов и синглтоны) путем идентификации генного объекта, которому были назначены гены, на основе по описанной выше процедуре.Каждый генный объект связан ровно с одним филогенетическим профилем P. Сначала определяется, какая часть, F pw , филогенетических профилей n f идентична среди всех возможных сравнений между всеми объектами генов n f , принадлежащими к метаболический класс или путь, вычисляемый как:
Для каждого результирующего набора из n и ферментных генов, связанных с одним конкретным метаболическим классом или путем, был определен связанный набор из n f генных объектов (семейства генов и синглтоны) путем идентификации генного объекта, которому были назначены гены, на основе по описанной выше процедуре.Каждый генный объект связан ровно с одним филогенетическим профилем P. Сначала определяется, какая часть, F pw , филогенетических профилей n f идентична среди всех возможных сравнений между всеми объектами генов n f , принадлежащими к метаболический класс или путь, вычисляемый как:
, где δ — дельта-функция Кронекера, дающая 1 в случае идентичных филогенетических профилей P i и P j .Профили считались идентичными, если они имели точно такой же битовый вектор, указывающий на присутствие и отсутствие во всех 39 рассматриваемых видах растений. Аналогично, фракция, F all , идентичных профилей среди всех n f_all генных объектов, связанных со всеми ферментными генами, независимо от метаболического класса или назначения пути с n f_all = n f + n allO , где n allO — количество генных объектов, не отнесенных к классу пути или тестируемому пути, вычисляется как:
Падение = ∑inf∑j = i + 1nfδPi, Pj + ∑i = 1nf∑j = 1, nfallOδPi, Pjnf (nf-1) 2 + nf * nfallO.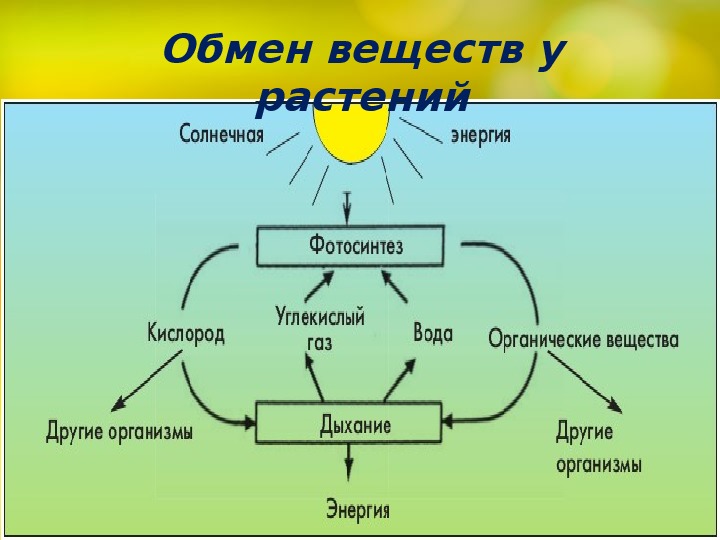 (2)
(2)Обратите внимание, что сравнения или филогенетические профили в уравнении (2) выполняются только для тех профилей, которые являются частью определенного метаболического класса или пути. Отношение F pw к F все дает обогащение, E = F pw / F все , идентичных профилей в наборе генных объектов пути относительно всех семейств генов в наборе данных. Обратите внимание, что в уравнении 2 два сравнения наборов объединены, появляясь как слагаемые в знаменателе и ограничителе, соответственно: сравнение внутри класса / профиля пути и сравнение со всеми другими внешними профилями.
Эмпирические p -значения оценки обогащения были вычислены путем случайного извлечения того же количества семейств генов, которое первоначально аннотировалось как принадлежность к определенному метаболическому классу или пути из всех генных объектов, и выполнения вычисления оценок обогащения для 10000 таких случайных прогонов, в результате средние случайные значения F pw , F all и связанное обогащение E r .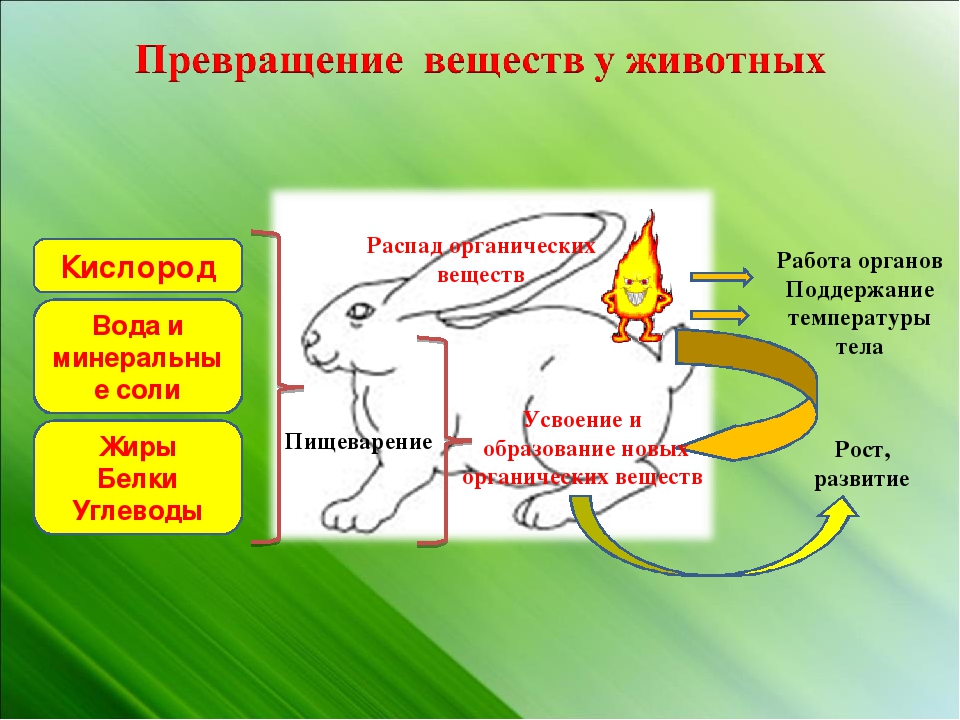 Было вычислено эмпирическое значение p , обозначающее долю равных или более высоких оценок обогащения, полученных в 10 000 случайных испытаний, чем для фактического набора генных объектов пути.Полученные значения p были скорректированы для многократного тестирования — столько же метаболических классов или путей было проверено — с помощью поправки Бенджамини-Хохберга, реализованной в функции p.adjust R.
Было вычислено эмпирическое значение p , обозначающее долю равных или более высоких оценок обогащения, полученных в 10 000 случайных испытаний, чем для фактического набора генных объектов пути.Полученные значения p были скорректированы для многократного тестирования — столько же метаболических классов или путей было проверено — с помощью поправки Бенджамини-Хохберга, реализованной в функции p.adjust R.
Тест на предсказуемость ассоциации путей на основе филогенетических профилей
Для проверки предсказательной силы сходства филогенетических профилей двух генных объектов в отношении ассоциации их путей были реализованы следующие две процедуры.Во-первых, мы проверили, приводит ли повышенное сходство филогенетического профиля между генами к увеличению вероятности того, что оба гена участвуют в одном и том же ферментативном пути. Во-вторых, мы использовали подход машинного обучения, чтобы проверить, можно ли предсказать членство в пути для данного отдельного гена непосредственно на основе только его филогенетических профилей. Более подробно, в первом подходе, повторенном 100000 раз, два генных объекта, g 1 и g 2 , были выбраны случайным образом из набора из всех 2206, включая семейства генов и синглтоны.Филогенетические профили PP 1 и PP 2 , каждый из которых представляет собой 39-элементный вектор, состоящий из единиц (присутствие в виде) и нулей (отсутствие вида), связанных с g 1 и g 2 , соответственно, сравнивались по их индексу Жаккара, измеряющему пересечение по сравнению с объединением записей «1» и их расстоянием, d PP , определенным как:
Более подробно, в первом подходе, повторенном 100000 раз, два генных объекта, g 1 и g 2 , были выбраны случайным образом из набора из всех 2206, включая семейства генов и синглтоны.Филогенетические профили PP 1 и PP 2 , каждый из которых представляет собой 39-элементный вектор, состоящий из единиц (присутствие в виде) и нулей (отсутствие вида), связанных с g 1 и g 2 , соответственно, сравнивались по их индексу Жаккара, измеряющему пересечение по сравнению с объединением записей «1» и их расстоянием, d PP , определенным как:
где «&» и «|» — побитовые операции И и ИЛИ, соответственно, и || 1 — L1-норма; я.е., сумма всех единиц в PP-векторах длиной 39. Для обоих генных объектов, g 1 и g 2 , были определены все карты путей KEGG, к которым аннотированы их гены-члены, и согласование, A_PW g1, g2 , между обоими списками путей, измеренными в соответствии с логикой индекса Жаккара:
A_PWg1, g2 = PWg1∩PWg2min (N1, N2), (4), где PW g 1 и PW g 2 — это списки путей, связанных с генными объектами g 1 и g 2 , соответственно, N N 1/2 — это номера различных путей в PW g 1 и PW g 2 , пересечение представляет собой количество путей, найденных в обоих списках путей.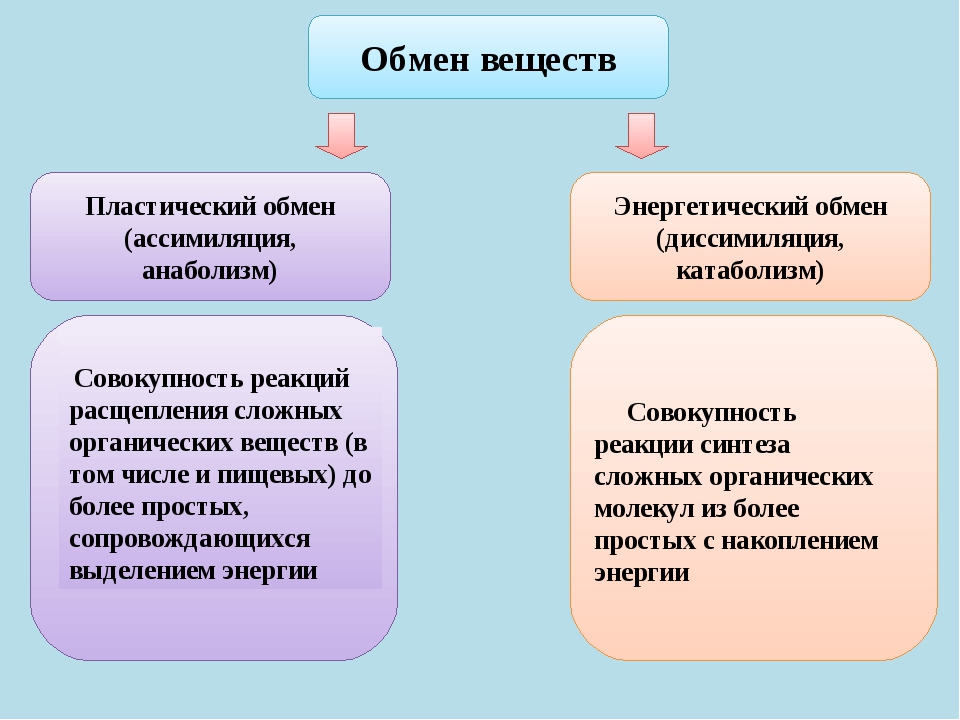 Обратите внимание, что мы намеренно решили брать выборку на основе генных объектов, а не на основе отдельных генов, поскольку последние могут смещать результат в пользу больших семейств генов.
Обратите внимание, что мы намеренно решили брать выборку на основе генных объектов, а не на основе отдельных генов, поскольку последние могут смещать результат в пользу больших семейств генов.
Пары значений d PP и A_PW g1, g2 из всех случайных испытаний были нанесены в виде диаграммы разброса (рисунок 7) и логистической функции A_PW = f (d PP ) с A_PW = 1 / (1 + exp (-a * (d PP — b))) был подогнан к данным с использованием нелинейной функции аппроксимации «nls» для R, а параметры a и b должны определяться с помощью аппроксимации.Была выбрана логистическая функция, поскольку она естественным образом сходится к нулю и единице, двум возможным крайним значениям A_PW.
Для подхода машинного обучения программное обеспечение Clus-HMC (Schietgat et al., 2010; Skunca et al., 2013) использовалось для прогнозирования класса метаболического пути или подробного пути (карта KEGG) для каждого генного объекта (семейства генов или singleton gene) на основе его филогенетического профиля. Пакет Clus-HMC идеально подходит, поскольку он позволяет использовать объекты с несколькими метками (объект гена и его функция могут быть назначены более чем одной карте путей) и потому, что он имеет дело с иерархическими данными (класс метаболизма и отдельные карты KEGG в качестве нижних уровень).Clus-HMC использует деревья решений в качестве механизма классификации. Мы использовали его в режиме случайного леса с 50 деревьями на прогон, метриками расстояния Жаккара и результатами прогнозирования, указанными на примерах вне пакета; то есть внутренняя перекрестная проверка, обычно используемая как часть методологии случайного леса. Эффективность оценивалась по площади под кривой точности-отзыва (AUCPRC), где точность определяется как отношение истинно положительного прогноза к сумме истинно положительного и ложноположительного прогнозов; я.е., какая доля из всех сделанных прогнозов верна. Отзыв определяется как отношение истинно положительных прогнозов к сумме истинно положительных и ложно отрицательных прогнозов; то есть, какая доля из всех положительных примеров в наборе данных была получена как положительные прогнозы.
Пакет Clus-HMC идеально подходит, поскольку он позволяет использовать объекты с несколькими метками (объект гена и его функция могут быть назначены более чем одной карте путей) и потому, что он имеет дело с иерархическими данными (класс метаболизма и отдельные карты KEGG в качестве нижних уровень).Clus-HMC использует деревья решений в качестве механизма классификации. Мы использовали его в режиме случайного леса с 50 деревьями на прогон, метриками расстояния Жаккара и результатами прогнозирования, указанными на примерах вне пакета; то есть внутренняя перекрестная проверка, обычно используемая как часть методологии случайного леса. Эффективность оценивалась по площади под кривой точности-отзыва (AUCPRC), где точность определяется как отношение истинно положительного прогноза к сумме истинно положительного и ложноположительного прогнозов; я.е., какая доля из всех сделанных прогнозов верна. Отзыв определяется как отношение истинно положительных прогнозов к сумме истинно положительных и ложно отрицательных прогнозов; то есть, какая доля из всех положительных примеров в наборе данных была получена как положительные прогнозы. Большие значения AUCPRC указывают на лучшие прогнозы. Поскольку наши данные сильно несбалансированы — для любого данного семейства генов им будет назначен только один или несколько путей из всех возможных — более известная область под ROC (истинно положительный vs.количество ложноположительных результатов) может ввести в заблуждение (Davis and Goadrich, 2006). В общей сложности 2206 генных объектов (семейства генов и одиночные гены), связанных с 816 уникальными филогенетическими профилями, были протестированы для отнесения либо к 10 классам метаболических путей, либо к 94 детальным картам путей. Значения AUCPRC, полученные для истинных ассоциаций генных объектов и их филогенетических профилей с классами и картами метаболических путей, сравнивались со значениями AUCPRC, полученными для рандомизированных назначений путем случайного перераспределения 10 классов метаболических путей и 94 карт путей для всех генных объектов, сохраняя при этом их появление и избегая повторяющиеся назначения генного объекта одному и тому же классу пути или карте.
Большие значения AUCPRC указывают на лучшие прогнозы. Поскольку наши данные сильно несбалансированы — для любого данного семейства генов им будет назначен только один или несколько путей из всех возможных — более известная область под ROC (истинно положительный vs.количество ложноположительных результатов) может ввести в заблуждение (Davis and Goadrich, 2006). В общей сложности 2206 генных объектов (семейства генов и одиночные гены), связанных с 816 уникальными филогенетическими профилями, были протестированы для отнесения либо к 10 классам метаболических путей, либо к 94 детальным картам путей. Значения AUCPRC, полученные для истинных ассоциаций генных объектов и их филогенетических профилей с классами и картами метаболических путей, сравнивались со значениями AUCPRC, полученными для рандомизированных назначений путем случайного перераспределения 10 классов метаболических путей и 94 карт путей для всех генных объектов, сохраняя при этом их появление и избегая повторяющиеся назначения генного объекта одному и тому же классу пути или карте.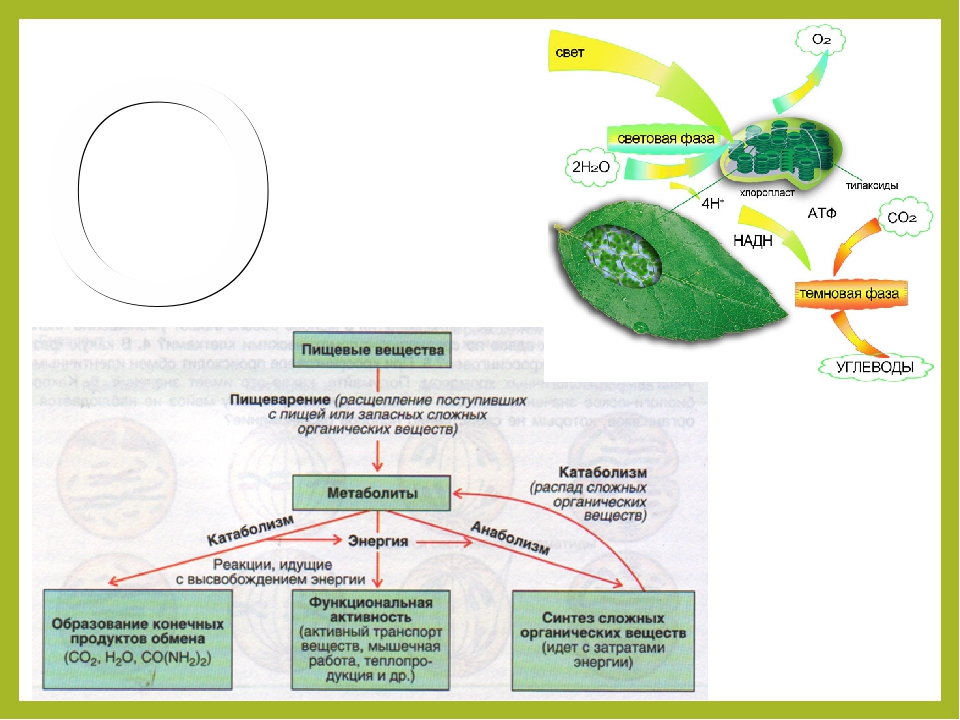 Этот процесс рандомизации был повторен 100 раз для предсказаний классов путей и 20 раз для карт путей. Для последнего потребовалось меньше случайных прогонов, поскольку их количество (94 карты против 10 классов) было намного больше. Статистические сравнения истинных и случайных предсказаний были выполнены с использованием непараметрического критерия суммы рангов Вилкоксона и усреднены по всем выполненным повторным запускам рандомизации.
Этот процесс рандомизации был повторен 100 раз для предсказаний классов путей и 20 раз для карт путей. Для последнего потребовалось меньше случайных прогонов, поскольку их количество (94 карты против 10 классов) было намного больше. Статистические сравнения истинных и случайных предсказаний были выполнены с использованием непараметрического критерия суммы рангов Вилкоксона и усреднены по всем выполненным повторным запускам рандомизации.
Сходство филогенетического профиля как показатель коэкспрессии генов и белок-белковых взаимодействий
Сходство филогенетических профилей двух генных объектов было проверено на предмет информативности в отношении совместной регуляции экспрессии их генов и физических взаимодействий их кодируемых продуктов посредством белок-белковых взаимодействий с акцентом на Arabidopsis thaliana в качестве эталонного вида с учетом обширной экспериментальной информации доступны для этого модельного вида растений в отношении как экспрессии генов, так и белок-белковых взаимодействий. Информация об экспрессии генов была получена из NASCArray (Craigon et al., 2004), охватывающая широкий диапазон экспериментальных условий, проверенных примерно пятью тысячами экспериментов по экспрессии гена генного чипа ATh2 Affymetrix (гибридизации). Необработанные данные по экспрессии генов были преобразованы в логарифмически и нормализованы по квантилю, как описано в Korkuc et al. (2014). Чтобы сократить время вычислений, для анализа использовалось случайное подмножество образцов, отобранных с вероятностью 10% из исходного набора образцов NASCArray, в результате чего были получены данные об экспрессии 20 922 генов в 479 гибридизациях.Для всех возможных пар из 500 случайно выбранных генов ферментов Arabidopsis их филогенетические профили сходства, d PP , были нанесены на график против их парного коэффициента корреляции Пирсона, r GE , экспрессии генов в 479 образцах экспрессии генов. В общей сложности для 93 961 пары генов Arabidopsis была доступна как филогенетическая, так и информация об экспрессии обоих генов, образующих пару ферментов, что позволяло проверить, соответствует ли повышенное филогенетическое сходство повышенной корреляции экспрессии их генов.
Информация об экспрессии генов была получена из NASCArray (Craigon et al., 2004), охватывающая широкий диапазон экспериментальных условий, проверенных примерно пятью тысячами экспериментов по экспрессии гена генного чипа ATh2 Affymetrix (гибридизации). Необработанные данные по экспрессии генов были преобразованы в логарифмически и нормализованы по квантилю, как описано в Korkuc et al. (2014). Чтобы сократить время вычислений, для анализа использовалось случайное подмножество образцов, отобранных с вероятностью 10% из исходного набора образцов NASCArray, в результате чего были получены данные об экспрессии 20 922 генов в 479 гибридизациях.Для всех возможных пар из 500 случайно выбранных генов ферментов Arabidopsis их филогенетические профили сходства, d PP , были нанесены на график против их парного коэффициента корреляции Пирсона, r GE , экспрессии генов в 479 образцах экспрессии генов. В общей сложности для 93 961 пары генов Arabidopsis была доступна как филогенетическая, так и информация об экспрессии обоих генов, образующих пару ферментов, что позволяло проверить, соответствует ли повышенное филогенетическое сходство повышенной корреляции экспрессии их генов.
Физические взаимодействия белков арабидопсиса были получены из базы данных AtPIN (Brandao et al., 2009). Взаимодействия с экспериментальной поддержкой учитывались только в сумме 95 219 парных белок-белковых взаимодействий среди 14 995 уникальных белков, из которых 5 978 пар, образованных среди 2265 генов, содержались в данных функциональной аннотации, а также идентифицировали их как ферменты. Для всех пар ферментов, которые, как сообщается, физически взаимодействуют, мы определили их ассоциированное сходство филогенетического профиля, d PP , и сравнили полученное распределение с распределением значений d PP , связанных с парами ферментов, о взаимодействии которых не сообщалось.Статистическая значимость была установлена на основе непараметрического критерия суммы рангов Вилкоксона.
Результаты
Обоснование филогенетического профилирования постулирует, что гены, коллективно выполняющие определенную биологическую функцию, присутствуют в виде набора только у тех видов, у которых эта функция выполняется.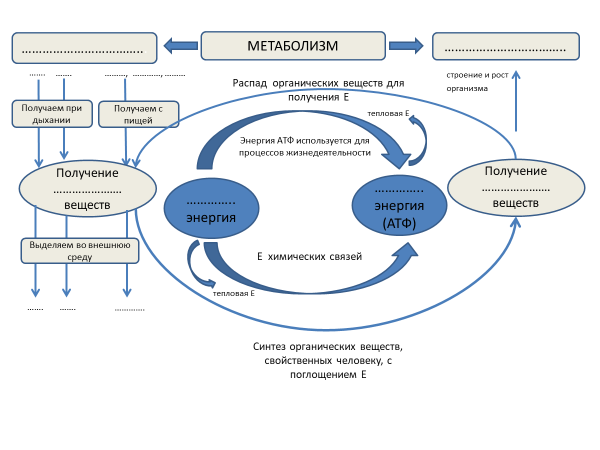 А для того, чтобы филогенетическое профилирование было конкретным, определенные функции должны быть связаны с уникальными филогенетическими профилями. Поскольку целью этого исследования было использование филогенетического профилирования генов для определения путей метаболизма с акцентом на вторичный метаболизм, мы сначала проверили наличие известных путей вторичного метаболизма у 24 видов растений с доступной информацией о Ensembl и KEGG (Рисунок 1).Первоначально пути считались присутствующими у определенного вида, если у этого вида был обнаружен хотя бы один ген, который был аннотирован как принадлежащий этому пути. На основании этого определения присутствия / отсутствия около одной трети (10 из 31 вторичного пути) было обнаружено во всех 24 видах растений. Таким образом, для этих путей не было очевидного дифференциального профиля присутствия / отсутствия, что делало бы применение филогенетического профилирования неспецифическим, поскольку ряд различных путей вторичного метаболизма демонстрируют один и тот же профиль присутствия.
А для того, чтобы филогенетическое профилирование было конкретным, определенные функции должны быть связаны с уникальными филогенетическими профилями. Поскольку целью этого исследования было использование филогенетического профилирования генов для определения путей метаболизма с акцентом на вторичный метаболизм, мы сначала проверили наличие известных путей вторичного метаболизма у 24 видов растений с доступной информацией о Ensembl и KEGG (Рисунок 1).Первоначально пути считались присутствующими у определенного вида, если у этого вида был обнаружен хотя бы один ген, который был аннотирован как принадлежащий этому пути. На основании этого определения присутствия / отсутствия около одной трети (10 из 31 вторичного пути) было обнаружено во всех 24 видах растений. Таким образом, для этих путей не было очевидного дифференциального профиля присутствия / отсутствия, что делало бы применение филогенетического профилирования неспецифическим, поскольку ряд различных путей вторичного метаболизма демонстрируют один и тот же профиль присутствия.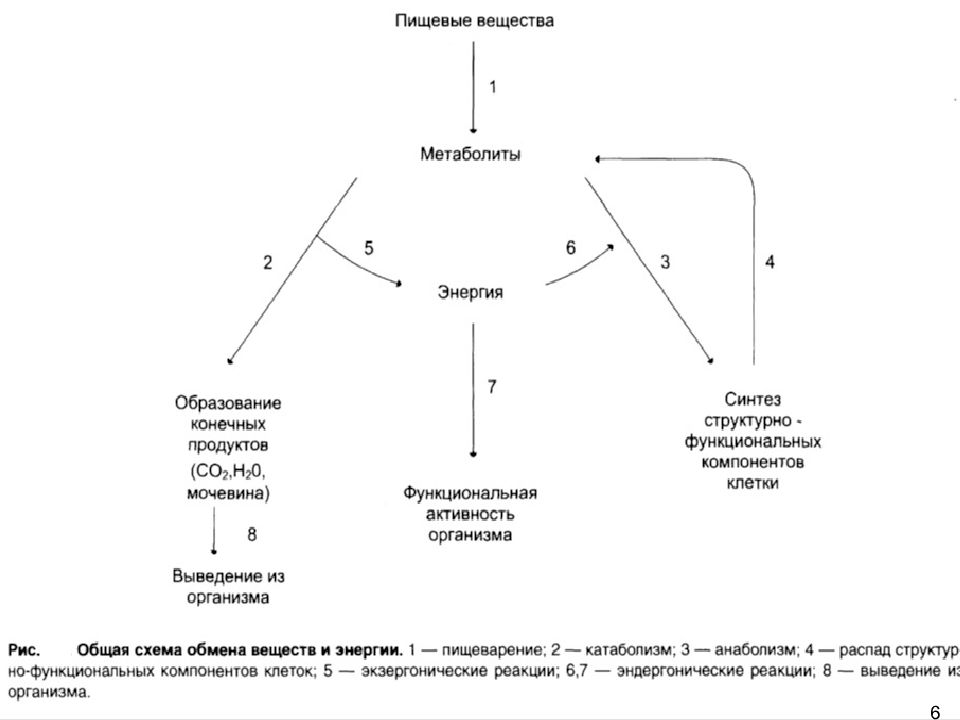 Очевидно, этот результат отражает текущую широту охвата видов, доступную в KEGG и Ensembl. Было обнаружено семь путей, присутствующих менее чем у половины всех видов KEGG, и еще 14 были обнаружены почти, но не у всех 24 видов (рис. 1). Таким образом, семь путей с ограниченным охватом видов кажутся наиболее многообещающими с точки зрения проверки филогенетического профилирования в качестве средства аннотации при условии, что их видовые спектры не сильно перекрываются. Они включают пути «биосинтеза пенициллина и цефалоспоринов», «биосинтез антибиотиков группы ванкомицина», «биосинтез карбапенема», «биосинтез изофлавоноидов», биосинтез бензоксазиноидов, «биосинтез индольных алкалоидов» и «биосинтез индольных алкалоидов» и «биосинтез индольных алкалоидов».Обратите внимание, что путь «биосинтез пенициллина и цефалоспоринов» указан в наборе данных о растениях, полученном от KEGG. Известно, что оба антибиотика вырабатываются грибами, но не растениями. Аннотации растений в KEGG происходят от фермента красных водорослей Cyanidioschyzon merolae , аннотированных как аналог D-аминокислотной оксидазы, которая, как известно, катализирует реакцию в пути биосинтеза пенициллина и цефалоспоринов, а также относится к основным путям метаболитов метаболизм аминокислот.
Очевидно, этот результат отражает текущую широту охвата видов, доступную в KEGG и Ensembl. Было обнаружено семь путей, присутствующих менее чем у половины всех видов KEGG, и еще 14 были обнаружены почти, но не у всех 24 видов (рис. 1). Таким образом, семь путей с ограниченным охватом видов кажутся наиболее многообещающими с точки зрения проверки филогенетического профилирования в качестве средства аннотации при условии, что их видовые спектры не сильно перекрываются. Они включают пути «биосинтеза пенициллина и цефалоспоринов», «биосинтез антибиотиков группы ванкомицина», «биосинтез карбапенема», «биосинтез изофлавоноидов», биосинтез бензоксазиноидов, «биосинтез индольных алкалоидов» и «биосинтез индольных алкалоидов» и «биосинтез индольных алкалоидов».Обратите внимание, что путь «биосинтез пенициллина и цефалоспоринов» указан в наборе данных о растениях, полученном от KEGG. Известно, что оба антибиотика вырабатываются грибами, но не растениями. Аннотации растений в KEGG происходят от фермента красных водорослей Cyanidioschyzon merolae , аннотированных как аналог D-аминокислотной оксидазы, которая, как известно, катализирует реакцию в пути биосинтеза пенициллина и цефалоспоринов, а также относится к основным путям метаболитов метаболизм аминокислот.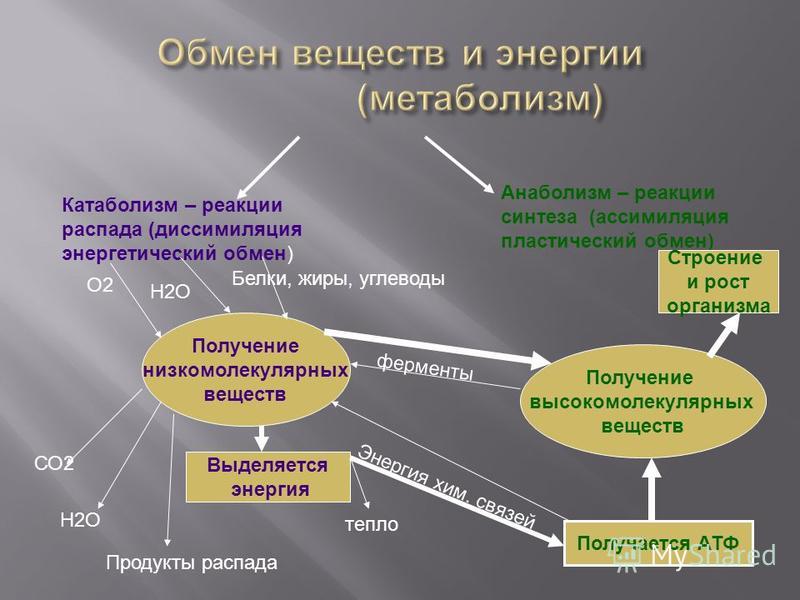 Поскольку этот путь фактически не осуществляется у растений, он не рассматривался в дальнейшем в этом исследовании. Аналогичным образом, бактериальный путь ванкомицина в дальнейшем также не рассматривался.
Поскольку этот путь фактически не осуществляется у растений, он не рассматривался в дальнейшем в этом исследовании. Аналогичным образом, бактериальный путь ванкомицина в дальнейшем также не рассматривался.
Рисунок 1. Статистика встречаемости путей вторичных метаболитов у 24 видов KEGG. Для каждого вторичного метаболического пути в базе данных KEGG отображается количество видов растений, содержащих его, из всех 24 видов растений, используемых в этом исследовании. Путь считался присутствующим у данного вида, если хотя бы один ген фермента этого вида был отнесен к этому пути.
Анализ распределения видов на путь был также выполнен для ферментов пути первичного метаболита, аннотированных в базе данных KEGG. Как и ожидалось для путей первичного метаболизма, поскольку они представляют важные функции, необходимые для выживания, большинство (71 из 81 пути первичного метаболизма, аннотированных в KEGG для 24 видов растений) были обнаружены у всех видов (данные не показаны).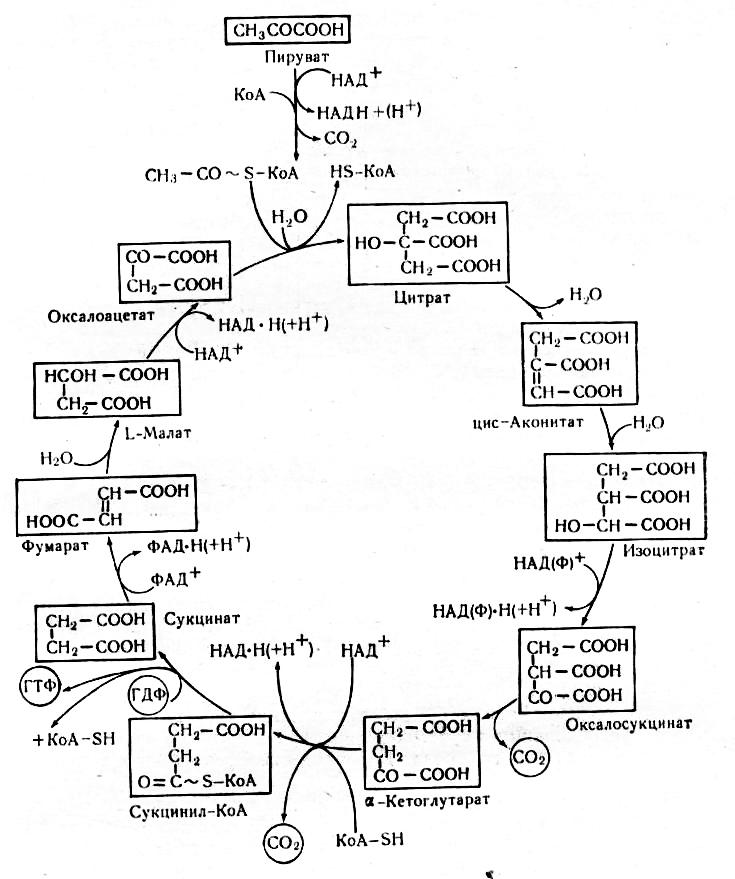
В исходном профиле присутствия / отсутствия путей вторичных метаболитов для всех 24 видов растений присутствие считалось подтвержденным, если был аннотирован хотя бы один компонентный ген, присутствующий в данном виде.Поскольку пути состоят из нескольких ферментов (в среднем 11 ферментов на путь вторичного метаболизма на основе набора данных, использованного в этом исследовании), и, кроме того, отдельные пути могут состоять из ветвей пути, действующих полунезависимо, более подробный анализ основан на пути гены-члены. Действительно, при проверке наличия отдельных генов-членов пути мы наблюдали, что, хотя отдельные гены-члены данного пути действительно были обнаружены у всех видов, другие гены, связанные с тем же путем, вполне могут иметь очень узкий спектр присутствия видов (рисунки 2, 3).Например, в пути «биосинтез дитерпеноидов» (номер карты KEGG 00904) около половины генов-членов были обнаружены у большинства видов, в то время как другая половина была обнаружена только у нескольких (одного) вида (рис.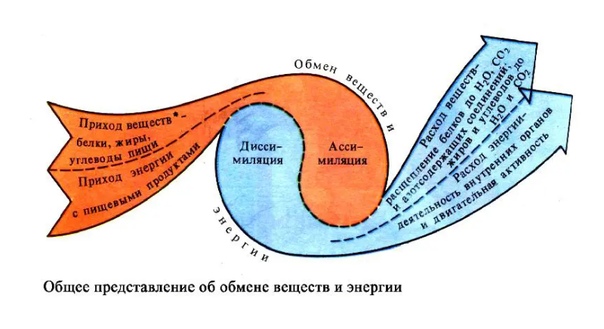 3). Путь «биосинтез сесквитерпеноидов и тритерпеноидов» (карта KEGG 00909) является еще одним примером ген-специфических профилей присутствия, связанных с одними и теми же путями. Напротив, для других путей было обнаружено, что все гены-члены встречаются практически у всех видов (например,g., «биосинтез терпеноидного остова», карта KEGG 00900 или «биосинтез флавоноидов», карта KEGG 00941, или последовательно только у нескольких видов (например, биосинтез изофлавоноидов, карта KEGG 00943).
3). Путь «биосинтез сесквитерпеноидов и тритерпеноидов» (карта KEGG 00909) является еще одним примером ген-специфических профилей присутствия, связанных с одними и теми же путями. Напротив, для других путей было обнаружено, что все гены-члены встречаются практически у всех видов (например,g., «биосинтез терпеноидного остова», карта KEGG 00900 или «биосинтез флавоноидов», карта KEGG 00941, или последовательно только у нескольких видов (например, биосинтез изофлавоноидов, карта KEGG 00943).
Рисунок 2. Подробные профили возникновения вторичных метаболитов. Для всех 31 пути вторичных метаболитов KEGG, указанных на их карте KEGG, присутствие на 24 видах растений, используемых в этом исследовании, нанесено на график для их соответствующих составляющих ферментов. Каждая полоса представляет собой фермент одного пути и их присутствие в 24 видах растений на основе аннотации идентичных номеров EC для разных видов.Ферменты сгруппированы и окрашены в соответствии с их номером карты KEGG.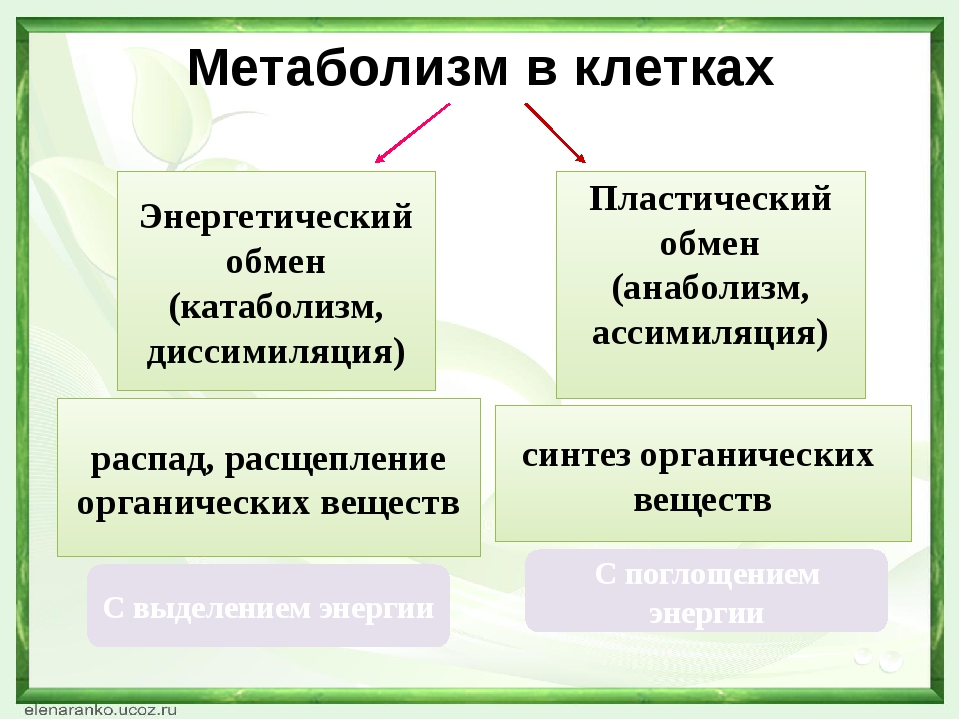 Названия связанных путей см. На Рисунке 1.
Названия связанных путей см. На Рисунке 1.
Рисунок 3. Карта пути KEGG пути биосинтеза дитерпеноидов (карта 00904). Все ферменты, выполняющие этапы основного линейного пути (выделены зеленым), были обнаружены как минимум в 19 видах растений. В отличие от этого, ветви путей, выделенные оранжевым цветом, были обнаружены только у одного или двух видов, а темно-красные ветви были обнаружены у 13 видов.Разрешение на воспроизведение этого изображения карты проезда было любезно предоставлено кураторами KEGG.
Подробные профили присутствия / отсутствия, показанные на Рисунке 2, уже показывают критическое ограничение филогенетического профилирования. Понятие коллективного присутствия или отсутствия не всегда может быть реализовано, учитывая нашу абстракцию изолированных биохимических путей, и может потребовать дальнейшего разделения отдельных биохимических реакций и функций. Это проиллюстрировано для пути биосинтеза дитерпеноидов (рис. 3).Хотя ферменты основной ветви этого пути были обнаружены почти у всех из 24 видов с доступной аннотацией пути, пути, ответвляющиеся от основного пути, присутствуют только у выбранных видов.
Число аннотированных генов, участвующих в пути, оказалось сильно варьирующим (рис. 4). Если рассматривать в качестве связанных генов только те гены, которые аннотированы исключительно для участия во вторичных, а не первичных путях метаболизма, не все из 31 вторичного пути метаболизма на самом деле содержат гены, при этом девять путей без каких-либо генов, специфичных для вторичного метаболического пути (например,г. «Метаболизм кофеина»), а другие содержат очень мало (например, «биосинтез антоциана»). Напротив, 13 путей имеют сотни («биосинтез флавоноидов», «биосинтез каротиноидов», «синтез дитерпеноидов») и даже тысячи («биосинтез фенилпропаноидов», «биосинтез терпеноидного остова») аннотированных им генов. Обратите внимание, что это количество генов включает в себя все ортологи и паралоги всех 24 видов растений, рассматриваемых здесь, и основано на аннотации номера EC, предоставленной KEGG.
Рисунок 4. Число генов, кодирующих ферменты, участвующие во вторичных метаболических путях. На графике нанесено количество генов, присвоенных каждому пути вторичных метаболитов растений, как указано в базе данных KEGG. Подсчет основан на генах всех растений любого из 24 видов растений, рассмотренных в этом исследовании. Числа вдоль оси абсцисс обозначают фактическое количество генов, участвующих во вторичных путях метаболизма, только цифры в скобках относятся к соответствующему количеству при рассмотрении аннотированных генов, участвующих как во вторичных, так и в первичных путях метаболизма.Пути обозначены их названиями. Пути «Биосинтез антибиотиков группы ванкомицина» как бактериальный путь и «биосинтез пенициллина и цефалоспорина» как грибковый путь больше не рассматривались в этом исследовании, ориентированном на растения. Для путей «Биосинтез бензоксазиноидов» и «Биосинтез изохинолиновых алкалоидов», перечисленных в KEGG, в Biomart содержались противоречивые аннотации или их отсутствие, и поэтому они были исключены из дальнейшего анализа. Все оставшиеся 19 путей вторичного метаболизма с ненулевым количеством генов KEGG были рассмотрены дополнительно.
Обобщая эти первоначальные данные обследования, становится очевидным, что, несмотря на большие объемы накопленных данных секвенирования и аннотации генома, фактические знания о вторичных путях в отношении видов и охвата генов, а также пригодность для тестирования подходов к филогенетическому профилированию относительно ограничены и ограничиваются только несколько путей вторичных метаболитов.
Оценка принадлежности генного семейства
Присвоение гомологии между всеми генами всех рассматриваемых видов растений является решающим шагом в филогенетическом профилировании, поскольку его результат напрямую определяет профиль присутствия-отсутствия определенных ферментативных активностей у рассматриваемых видов растений.
Мы действовали исходя из предположения, что все члены семейства генов выполняют только одну и одну функцию, а разные семейства генов выполняют разные функции. Чтобы проверить это предположение, мы сравнили присвоение генов семействам генов с функциональными назначениями, указанными в номере EC KEGG, с 994 различными идентификаторами ферментов KEGG в наборе данных, что указывает на множество различных ферментативных активностей. Наш сетевой подход с порогом идентичности последовательностей 30 процентов (Network30) привел к появлению 2206 генных объектов, включая 1686 семейств генов и 520 одноточечных генов, что привело к скорректированному индексу Рэнда, равному 0.471. Использование порога идентичности последовательностей в 70% дало 9 285 объектов (4 373 генных семейства и 4 912 одноточечных генов) и скорректированный индекс Рэнда 0,116. Таким образом, более обширная кластеризация, позволяющая группировать вместе последовательности с большим расхождением, дала лучшее согласие с фактическими назначениями биологических функций. Напротив, разделение генов на семейства генов с более высокими пороговыми уровнями идентичности последовательностей, по-видимому, не объединяет гены в кластер по сравнению с фактической функцией. Однако следует предупредить, что функциональное назначение KEGG, в свою очередь, может быть основано на сравнении последовательностей.Таким образом, две кластеризации не могут быть полностью независимыми.
Поскольку распределение семейств генов Network30 оказалось более согласованным с фактической биохимической функциональной аннотацией, мы использовали его впредь для тестирования методологии филогенетического профилирования для определения взаимосвязей между путями.
На рис. 5 показаны подсчеты отдельных генов, аннотированных как ферменты, связанные пропорции, присвоенные семействам генов и одиночным генам, соответственно, для всех 24 аннотированных KEGG видов растений.Примечательно, что количество одиночных генов не коррелирует с общим количеством ферментов у вида. Кроме того, три вида водорослей Chlamydomonas reinhardtii (CRE), Ostreococcus lucimarinus (OLU), Cyanidioschyzon merolae (CME) характеризуются выраженным пониженным числом генов, относимых к семействам, но пропорционально большим количеством генов-одиночек. вероятно, отражая их эволюционное расстояние от высших растений.
Рисунок 5. Число генов ферментов, отнесенных к семействам генов. Доля генов, отнесенных к семействам генов (светло-серый), а также к генам-одиночкам (темно-серый) отображается для каждого вида растений с доступными KEGG и аннотациями генов. Распределение показано для присвоения семейств генов на основе набора данных Network30. Виды отсортированы по общему количеству генов. Для каждого вида учитывались только гены, аннотированные как ферменты.
Филогенетическое профилирование
Обогащение идентичных профилей в рамках различных метаболических путей
Основная цель этого исследования состояла в том, чтобы оценить полезность филогенетического профилирования как средства связывания генов на основании идентичных профилей с общей биологической функцией, здесь метаболическим путем и, в частности, путем вторичного метаболизма.Если верно то, что соответствие филогенетического профиля подразумевает участие общего пути, то для любого данного известного пути должно быть более высокое, чем ожидалось случайным образом, соответствие филогенетических профилей, связанных с присвоенными ему генами (высокие значения F pw , уравнение 1). Поскольку высокая степень совпадения профилей может также просто отражать то, что соответствующие профили очень часто встречаются во всех генах и участвуют во многих различных путях, нам необходимо сравнить согласие внутри пути с ожидаемым совпадением на основе общей частоты этого филогенетический профиль (F все , уравнение 2).Обратите внимание, что, говоря о генах, мы фактически имеем в виду генные объекты, определенные как семейства генов или гены-одиночки, как описано выше. И каждый генный объект характеризуется филогенетическим профилем. Каждый реальный ген, присутствующий у данного вида, является представителем генного объекта, который выполняет определенную ферментативную функцию. Это предположение не совсем верно (скорректированный индекс Рэнда 0,471), но, тем не менее, составляет оперативную и разумную основу нашего подхода. Как указано выше, мы основали следующий анализ на назначении семейств генов на основе Network30, как описано в разделе «Методы».
Мы провели статистические тесты, рассматривая два уровня абстракции метаболических путей: очень грубый уровень классов метаболизма — в KEGG определены 10 различных классов метаболизма (таблица 1, обратите внимание, что мы не считали класс «Обзор» слишком общим) и более подробная функциональная группировка, отраженная как метаболические пути. Всего мы рассмотрели 94 различных карты путей, из которых 19 были помечены как вторичный метаболизм. Каждый путь принадлежит к определенному классу метаболизма KEGG (дополнительная таблица 1).
Для пяти из 10 рассмотренных метаболических классов действительно наблюдалось значительное обогащение (множественное тестирование, скорректированное на p — значение <0,05) филогенетических профилей генных объектов, аннотированных к одному и тому же классу, относительно случайного ожидания. Все пять относятся к классам первичного метаболизма и включают: «Метаболизм аминокислот (AAM)», «Метаболизм других аминокислот (MOAA)», «Метаболизм кофакторов и витаминов (MCV)», «Метаболизм нуклеотидов (NM)» и «Углеводный обмен (УМ)» (Таблица 2).Уровни кратного обогащения для двух классов вторичного метаболизма «Метаболизм терпеноидов и поликетидов (MTP)», «Биосинтез других вторичных метаболитов (BSM)», хотя и превышали единицу (1,435 и 1,268 соответственно), не оказались статистически значимый. Из трех оставшихся метаболических классов «энергетический метаболизм (ЭМ)» показал пограничное значительное обогащение (1,217-кратное, скорректированное значение p = 0,09), в то время как «метаболизм липидов (LM)» и «биосинтез и метаболизм гликанов (GBM) »Не показали заметного обогащения (Таблица 2).
Таблица 2. Статистика распределения классов метаболизма и идентичности филогенетических профилей семейств генов и одиночек.
Результат значительного согласия филогенетических профилей, связанных с первичными и, следовательно, повсеместными путями, поначалу кажется удивительным, учитывая, что мы утверждали, что филогенетическое профилирование идеально подходит для идентификации биохимических функций, ограниченных подмножествами видов. Тем не менее, это обоснование по-прежнему актуально и имеет смысл.Филогенетические профили, отражающие присутствие у всех видов, информативны в том смысле, что они идентифицируют функции, которые необходимы, и поскольку многие профили будут отражать присутствие только в подмножествах видов, даже те профили, которые предполагают присутствие у всех видов, могут быть обогащены относительно случайного ожидания. И они обогащены именно теми курсами подготовки, которые необходимы. Однако специфичность назначения путей может быть потеряна, так как многие различные пути будут важны и, следовательно, выполняются у всех видов (см. Рисунок 6 и связанные с ним результаты ниже).
Рис. 6. Статистика частоты филогенетического профиля. Для всех 818 уникальных филогенетических профилей — их встречаемость; то есть количество генных объектов с этим профилем отображается в порядке убывания встречаемости (A) . Соответствующее фактическое присутствие (черная клетка) / отсутствие (белая клетка) для всех 39 видов растений, рассмотренных в этом исследовании, нанесено на график под (B) . Виды растений даны по их аббревиатуре, представленной в Таблице 1, и сгруппированы иерархически (полная связь, Евклидово расстояние) в соответствии с сходством модели присутствия / отсутствия.Уникальные профили, найденные только для одного генного объекта, сортируются по убыванию числа генов, сгруппированных в этот объект; то есть от больших семейств генов (сгруппированных слева) до небольших семейств генов и одиночных генов (сгруппированных справа). Обратите внимание, что мы использовали только те объекты генов, для которых была доступна аннотация KEGG хотя бы для одного растения-члена.
На более детальном уровне функциональной абстракции с учетом фактических метаболических путей, как аннотировано в KEGG, 29 из 94 рассмотренных путей показали значительное обогащение (множественное тестирование скорректировано p -значение <0.05) идентичных филогенетических профилей между объектами генов-членов относительно случайного ожидания (Таблица 3). Опять же, следуя той же логике, которая была объяснена выше для метаболических классов, большинство из 29 путей относятся к основным путям метаболизма, таким как цикл TCA (кратное обогащение, E = 4,145, скорректированное значение p <0,001) или различные пути метаболизма аминокислот (таблица 3). Однако наибольшие, а также значительные факторы обогащения наблюдались для двух вторичных путей метаболизма: «Биосинтез стилбеноидов, диарилгептаноидов и гингерола» ( E = 12.553, p = 0,024) и погранично значимые пути «биосинтез дитерпеноидов» ( E = 11,626, p = 0,069) и «биосинтез флавоноидов» ( E = 4,695, p = 0,097). Из оставшихся 17 путей вторичного метаболизма 11 содержали три или меньше различных генных объектов (дополнительная таблица 2), что делало невозможным какую-либо значимую статистическую оценку. Обратите внимание, что мы рассматривали только генные объекты как относящиеся к вторичным путям метаболизма, которые также не участвуют в процессах первичного метаболизма.Следовательно, количество генных объектов может быть значительно меньше, чем количество ферментов, аннотированных в KEGG, как принадлежащих к конкретному вторичному пути метаболизма. Путь вторичного метаболизма «деградация лимонена и пинена» демонстрирует высокое обогащение профильных соглашений ( E = 7,296), но статистическую значимость установить не удалось ( p = 0,142). Все другие пути вторичного метаболизма («биосинтез терпеноидного остова», «биосинтез каротиноидов», «биосинтез фенилпропаноидов», «биосинтез зеатина») не показали заметного обогащения профильных соглашений внутри них, несмотря на относительно большое количество назначенных им генных объектов (> 10). (Дополнительная таблица 2).
Таблица 3. Статистика назначений путей метаболизма и идентичности филогенетических профилей семейств генов и одиночек.
Мы рассматривали как потенциально функционально связанные только те генные объекты, которые имеют один и тот же филогенетический профиль. В то время как более мягкие пороговые значения (допускающие небольшое количество несоответствий наличия / отсутствия среди 39 рассматриваемых видов растений) или даже постепенные расстояния между профилем и профилем, основанные на битовых расстояниях, возможны, учитывая используемый здесь критерий « все или ничего », проверяющий частоту Распределение уникального филогенетического профиля может пролить дополнительный свет на представление уникальных профилей во всех семействах генов (Рисунок 6).Всего мы определили 818 уникальных профилей, связанных с 2206 объектами генов (семейства генов или гены-одиночки). Профиль присутствия во всех видах наблюдался наиболее часто. С помощью этого профиля было обнаружено сто восемьдесят генных объектов. Было обнаружено, что 131 профиль является общим для двух или более генных объектов, тогда как было обнаружено, что 685 профилей однозначно связаны только с одним генным объектом (рис. 6). Уникальные профили — это как профили, которые характеризуются присутствием только у небольшого числа видов, так и общее присутствие с уникальными отсутствием у отдельных видов.На рисунке 6 также показаны профили присутствия / отсутствия для 39 видов растений, рассмотренных в этом исследовании. Поскольку это наиболее широко и интенсивно исследуемая модель растения, A. thaliana выделяется как обладающая наибольшим количеством вызовов присутствия. Поскольку аннотация генома других видов также часто происходит от Arabidopsis на основе сравнения последовательностей, другие виды могут по существу обладать только меньшим, но не большим количеством функционально аннотированных объектов генов, если только не будут исследованы более тщательно и экспериментально или основаны на аннотации биоинформатических генов de-novo.Группирование видов растений в соответствии с сходством профиля их присутствия / отсутствия по всем 818 уникальным профилям воспроизводит установленные филогенетические отношения между ними (рис. 6).
Предсказуемость ассоциации метаболических путей на основе сходства филогенетических профилей
Мы наблюдали, что для выбранных классов путей (таблица 2) и детальных путей (таблица 3) действительно очевиден значительный рост встречаемости генов с тем же филогенетическим профилем.Мы теперь спросили, верно ли обратное, высокое сходство, подразумевает ли связь с одним и тем же путем. В то время как первое можно рассматривать как необходимое условие для того, чтобы филогенетические профили имели прогностическую ценность, последнее представляет собой окончательный тест и определяет применимость филогенетического профилирования на практике. Изменение точки зрения на противоположное (ассоциация путей предполагает сходство профилей, а сходство профилей предсказывает ассоциацию путей) также не эквивалентно из-за общего отсутствия симметрии условной вероятности двух событий A и B с P (A | B) ≠ P (B | А) в большинстве случаев.
Чтобы оценить прогностическую ценность сходства филогенетических профилей в отношении отнесения двух генов к одному или разным путям, мы случайным образом извлекли два разных генных объекта из всех 2206 генных объектов, включающих 1686 семейств генов и 520 одноэлементных генов, и определили их сходство с относительно филогенетического профиля и назначений путей (см. раздел «Материалы и методы»). Если предсказуемо, высокое сходство филогенетических профилей, отраженное в нашем подходе значениями d PP (уравнение 3), близкими к единице, должно указывать на высокое совпадение назначений путей двух генных объектов с приближением значений A_PW (уравнение 4) к единице.Однако, как показано на рисунке 7, мы не наблюдали корреляции между обоими показателями сходства, что позволяет нам сделать вывод, что с учетом имеющихся данных и применяемых определений сходство филогенетического профиля не является предиктором ассоциации путей.
Рисунок 7. Метаболический путь — статистика корреляции филогенетического профиля. Попарное согласие членства в путях, A_PW, генных объектов (семейств и одиночек) относительно сходства их соответствующих филогенетических профилей, d PP .Поскольку базовые данные дискретны, что приводит к множеству идентичных пар значений, их соответствующая частота показана областью кружков с центром на наблюдаемых парах значений. Красная линия означает логистическое соответствие (см. Раздел «Материалы и методы») и предполагает, что сходство путей не может быть выведено на основании сходных филогенетических профилей пар генов.
Затем мы рассматривали задачу назначения путей на основе филогенетических профилей как проблему машинного обучения.Использование программного пакета Clus-HMC (подробности см. В разделе «Материалы и методы»), который позволяет использовать иерархические структуры данных в качестве целей прогнозирования (класс пути с подробными картами путей на следующем более низком уровне), а также позволяет использовать несколько меток (ген может участвовать в более одного пути), мы стремились предсказать метаболический класс и карту путей для всех семейств генов и генов-одиночек на основе их филогенетического профиля. Подход к прогнозированию с помощью методологии машинного обучения (случайные леса), возможно, позволит выбранным видам получить более высокую прогнозную ценность, чем обработка всех записей одинаково, как это делается в метрике сравнения профиля и профиля.В настройке перекрестной проверки (ошибка вне пакета в предсказаниях дерева классификации случайных лесов) наивысшая точность прогноза была достигнута для центральных путей метаболизма с учетом класса метаболизма (таблица 4, рисунок 8) и подробных карт путей «Фотосинтез, «Взаимопревращения пентозы и глюкуроната», «Метаболизм крахмала и сахарозы», «Метаболизм пиримидина» и «Пуриновый метаболизм» (таблица 5), опять же пути, связанные с первичным метаболизмом. По сравнению со случайно перетасованными данными на уровне карты путей были получены значительно лучшие, чем случайные предсказания ( p = 0.0033, рисунок 8), тогда как для данных на уровне класса значимость не могла быть установлена, хотя правильные данные соответствовали большим областям под кривой точности-отзыва (AUCPRC), чем полученные для перетасованных данных ( p = 0,22, рисунок 8) .
Таблица 4. Результаты предсказания случайного леса Clus-HMC метаболического класса на основе филогенетического профиля генных семейств и одиночных генов.
Рисунок 8. Результаты классификации. Результаты классификации классов метаболических путей или сопоставления объектов генов на основе их филогенетических профилей с использованием прогнозов случайного леса, реализованных в Clus-HMC.Производительность оценивается по площади под кривой точности-отзыва (AUCPRC). Для 100/20 рандомизированных повторов, выполненных для карты пути или класса соответственно, нанесены средние распределения AUCPRC. Усредненные по всем случайным повторным запускам, тесты на статистическую разницу (критерий суммы рангов Уилкоксона) между фактическим и случайным распределениями значений AUCPRC дали для класса пути: среднее фактическое = 0,2, среднее случайное = 0,13, p = 0,22, и для карты путей: среднее фактическое = 0,04, среднее случайное = 0,021, p = 0.0033. Использовался Clus-HMC, позволяющий множественные и иерархически организованные метки для каждого объекта с иерархией, связанной с классом метаболизма и картой метаболизма.
Таблица 5. Результаты предсказания случайного леса Clus-HMC карты метаболических путей, основанные на филогенетическом профиле семейств генов и одиночных генов.
Сходство филогенетического профиля как показатель коэкспрессии генов и белок-белковых взаимодействий
До сих пор мы стремились сделать выводы о взаимоотношениях генов в метаболических путях на основе сходства их филогенетических профилей.Затем мы исследовали, оказываются ли филогенетические профили информативными в отношении регуляции коэкспрессии генов кодируемых транскриптов и физических взаимодействий их белковых продуктов. Подобно членству в метаболических путях, оба типа ассоциаций можно рассматривать как свидетельство участия в сходных функциональных процессах (Durek and Walther, 2008; Walther et al., 2010). Поскольку физические взаимодействия (белок-белковые взаимодействия) представляют собой прямые ассоциации, обоснование сходства филогенетических профилей, отражающих функциональные метаболические ассоциации, может стать наиболее очевидным при их тестировании на сравнении белок-белковых взаимодействий.В этом анализе мы сосредоточили внимание на генах растения A. thaliana , поскольку имеется обширная экспериментальная информация об экспрессии генов и белок-белковых взаимодействиях. В частности, мы выбрали пары генов, кодирующих ферменты, из Arabidopsis, извлекли профили их экспрессии из NASCArray, коррелировали их, а также проверили, сообщалось ли о взаимодействии их белковых продуктов (подробности см. В разделе «Материалы и методы»). Затем мы проверили, правильно ли отражается их ассоциация, судя по совместной экспрессии или физическому взаимодействию, сходством филогенетических профилей семейств генов, к которым принадлежат гены Arabidopsis.Поскольку это исследование сосредоточено на метаболических аспектах, мы рассматривали только гены, кодирующие ферменты.
Что касается коэкспрессии генов метаболических ферментов арабидопсиса, не было обнаружено никаких доказательств увеличения сходства профилей, что отражается усилением регуляции коэкспрессии ( r = 0,022, p = 2,9 * 10 −11 , рис. 9), хотя корреляция между двумя измерениями расстояния оказалась значительной, но из-за большого количества пар значений. Напротив, явная и статистически значимая разница была обнаружена при тестировании белок-белковых взаимодействий.Было обнаружено, что физически взаимодействующие ферменты связаны с генами, филогенетические профили которых более похожи друг на друга (среднее значение d PP = 1), чем для невзаимодействующих ферментных белков (среднее значение d PP = 0,92, p <2,2 * 10 −16 , рисунок 10). Таким образом, как утверждалось выше, прямые взаимодействия действительно отражаются сходством филогенетических профилей, в то время как коэкспрессия генов, которая включает пары генов, которые действуют в отдаленных функциональных процессах, обычно нет.
Рисунок 9. Статистика парной коэкспрессии генов. Ассоциация попарного сходства филогенетического профиля, d PP , и совместной экспрессии пар генов Arabidopsis, о чем судят по их парному коэффициенту корреляции Пирсона, r GE . Необработанные попарные данные показаны в виде пустых кружков, красная линия указывает на линию линейной регрессии ( r = 0,022, p = 2,9 * 10 −11 ), зеленая линия соответствует логистической подгонке, практически совпадающей с линейной линия регрессии, синие кружки обозначают медианные значения данных с интервалом (ширина интервала = 0.1 d PP шт.) Соединены прямыми линиями для визуального наведения.
Рисунок 10. Статистика белок-белкового взаимодействия. Скрипичный график частотного распределения значений сходства филогенетических профилей, d PP , пар ферментов, кодируемых в Arabidopsis thaliana , которые, как сообщается, физически взаимодействуют (медиана = 1) с теми парами, которые, как сообщается, не взаимодействуют (медиана = 0,92, p <2,2 * 10 −16 ).
Обсуждение
В этом исследовании мы проверили применимость сходства филогенетических профилей в качестве индикатора функциональной ассоциации между генами.В частности, мы стремились определить, могут ли паттерны коррелированного присутствия или отсутствия генов и их конкретных функций в разных геномах растений использоваться для вывода взаимосвязей метаболических путей. Мы сосредоточились на вторичном метаболизме, поскольку известно, что вторичный метаболизм проявляет явную привязку к определенным видам, что является предпосылкой для информативности филогенетического профилирования и, таким образом, успешного прогнозирования функциональных ассоциаций генов. Мы выполнили ряд анализов на основе 39 геномов растений и функциональной аннотации генов, доступных в KEGG.Мы подошли к ответу на ключевые вопросы этого исследования с разных сторон, сначала проверив, демонстрируют ли гены, относящиеся к известным метаболическим путям, большее, чем ожидалось, соответствие между их филогенетическими профилями. Затем мы изменили этот подход, задав вопрос, можно ли предсказать ассоциацию путей на основе филогенетических профилей. Наконец, мы также проверили, влияет ли сходство филогенетического профиля на коэкспрессию генов и физические взаимодействия их кодируемого белкового продукта.
Что касается центрального вопроса о взаимосвязи метаболических путей, результаты нашего технико-экономического обоснования были в основном отрицательными.Сходство филогенетических профилей не оказалось информативным в отношении взаимоотношений метаболических путей. Точнее, никаких конкретных прогнозов сделать не удалось. Правильные ассоциации путей были предсказаны только для основных путей метаболизма (Рисунок 8, Таблицы 2–5). Таким образом, методология правильно предсказала основные процессы на основе вызовов наличия / отсутствия генов в геномах, которые, однако, включают несколько путей. Таким образом теряется специфика. Установление взаимосвязей вторичных путей оказалось невозможным, а коэкспрессия генов не коррелировала со сходством филогенетических профилей (рис. 9).Единственным, но очень заметным исключением были физические взаимодействия между белковыми продуктами (рис. 10). Здесь филогенетические профили доказали свою прогностическую ценность. Прямые физические взаимодействия ферментных белков отражаются повышенным сходством филогенетического профиля кодирующих их генов. Таким образом, вместо того, чтобы распространяться на ассоциации генов на больших функциональных расстояниях, захваченных назначением генов на общий путь, филогенетические профили оказались предсказуемыми только для краткосрочных, фактически прямых функциональных взаимодействий, включающих физические контакты.
В значительной степени отрицательные результаты этого технико-экономического обоснования требуют критического анализа дизайна исследования, используемых данных и применяемой методологии. Критические аспекты касаются выбора видов растений, отнесения генов к семействам генов в качестве критического шага к установлению наличия или отсутствия определенных функций генов в геномах, а также разнообразия и точности аннотаций метаболических путей, особенно в отношении вторичных путей метаболизма. как понятие различных метаболических путей как подходящая абстракция функциональных взаимодействий между ферментами.
Выбор видов растений
По сути, подход филогенетического профилирования основан на корреляциях векторов (бинарные вызовы наличия / отсутствия генов или их соответственно кодируемых функций). Таким образом, как и в случае любой меры корреляции, достоверность значимой корреляции должна возрастать с увеличением длины векторов; то есть количество пар значений, или геномов в нашем случае, для сравнения. Поэтому представляется желательным включить как можно больше геномов / видов растений. При тестировании необходимого минимального количества геномов для включения в исследования филогенетического профилирования результаты прогнозов выходили на плато, превышая 100 включенных геномов, с дополнительной важностью, связанной с увеличением филогенетического разнообразия, а не с голым подсчетом геномов (Škunca and Dessimoz, 2015).Точно так же сообщалось, что помимо включения многих геномов, их отбор (например, в отношении царства) также имеет значение, и что, кроме того, отбор геномов может по-разному влиять на предсказуемость различных путей (Jothi et al., 2007).
Сюда мы включили 39 видов растений, охватывающих широкий спектр видов растений, от водорослей до высших растений (Таблица 1). Несмотря на то, что количество геномов является сравнительно многообещающим, если судить по описанным 100 видам оптимального, очевидно, что дальнейшее увеличение этого количества видов было бы желательным, но это зависит от наличия секвенированных геномов растений, которые, вероятно, будут быстро расти с учетом прогресса в технологии секвенирования.В качестве альтернативы мы могли бы увеличить количество рассматриваемых геномов, включив в него нерастительные виды. Однако, поскольку нас особенно интересовал вторичный метаболизм растений, который (в основном) отсутствует у нерастительных видов, мы отказались от него. Включение видов, не относящихся к растениям, вероятно, дало бы надежные прогнозы для определения взаимоотношений генов, присутствующих только в растениях. Тем не менее, мы полагаем, что различение генов и функций растений может быть достигнуто только на основе сравнения отдельных генов без навязывания коррелированных паттернов наследования, необходимых специально для выявления функциональных взаимосвязей.Включение нерастительных геномов привело бы к неспецифическим результатам, что взаимодействие ограничивается царством растений.
Отнесение генов к семействам генов как критический шаг к установлению наличия или отсутствия определенных функций генов в геномах
Наиболее важно то, что филогенетическое профилирование зависит от правильного определения присутствующего гена или его отсутствия. Точнее, необходимо решить, может ли конкретная функция, выполняемая у референсных видов, выполняться у других видов, и, следовательно, в ней будет закодирован гомологичный ген.
Мы действовали исходя из предположения, что сходные по последовательности ферменты выполняют схожие или идентичные функции. Следовательно, наличие или отсутствие определенной ферментативной активности в данном геноме может быть определено на основе сходства последовательности с аннотированным эталонным геном. Хотя было показано, что сходство последовательностей действительно является хорошим предиктором сходной структуры белка и, следовательно, функции (Sander and Schneider, 1991), и что функциональные различия идентичности последовательностей выше 40% маловероятны (Lo Conte et al., 2002; Orengo et al., 2002), также были описаны противоречащие друг другу примеры. Например, белки с высоким сходством последовательностей с генами, связанными с фотосинтезом, были обнаружены у нефотосинтетически активных организмов, что делает недействительным любое основанное на последовательностях функциональное назначение фотосинтетическим процессам (Ashkenazi et al., 2012). В более общем плане, ранние выводы, предполагающие относительно низкие пороги идентичности последовательностей как достаточные для надежной передачи функциональной аннотации, были поставлены под сомнение, указав на возможные систематические ошибки в базе данных (Rost, 2002).Продолжая это исследование, Тиан и Сколник показали, что при 40% идентичности последовательностей передача ферментативной функции на уровне первых трех цифр номера EC является надежной. Однако для предсказания всех четырех цифр необходимы 60% -ные уровни идентичности последовательностей для достижения 90% -ной точности (Tian and Skolnick, 2003). Таким образом, кажется удивительным, что мы получили наилучшее согласие кластеризации семейств генов и аннотации ферментативной функции при 30%, а не 70% идентичности последовательностей. Мы полагаем, что это очевидное противоречие объясняется осознанием того, что Тиан и Сколник (2003) исключили все вычислительные предсказания, в то время как мы включили их.Таким образом, наши результаты будут зависеть от пороговых значений сравнения последовательностей, применяемых кураторами исходного генома, которые, скорее всего, включали более общие пороговые уровни идентичности последовательностей. А поскольку наш набор данных о растениях содержит множество видов, которые менее интенсивно изучались экспериментально, вычислительные аннотации лягут в основу многих функциональных назначений. Кроме того, очевидно, что идентичность последовательности по всей последовательности может быть только в среднем хорошим предиктором функции, поскольку даже мутации одной аминокислоты может быть достаточно для изменения функции фермента, например, в отношении специфичности субстрата (Khersonsky et al., 2006).
Для генерации семейств генов мы также протестировали OrthoFinder (Emms and Kelly, 2015), а также применили алгоритмы выявления сообществ к сетям на основе сетей на основе сравнения последовательностей для выявления подкластеров генов, которые можно рассматривать как отдельные семейства генов. Однако эти попытки не привели к качественно отличным результатам, чем описанные здесь, на основании попарного определения пороговых значений идентичности последовательностей.
Обширность и точность аннотации метаболических путей
Мы специально стремились использовать филогенетическое профилирование для идентификации генов, обычно участвующих в конкретных путях вторичного метаболизма растений.Поскольку известно, что вторичные пути метаболизма происходят конкретно у определенных видов (Hartmann, 1996; Higashi and Saito, 2013), требования филогенетического профилирования кажутся идеально удовлетворенными. Однако для путей вторичного метаболизма были получены низкое статистическое соответствие профилей генов-членов (таблицы 2, 3, дополнительная таблица 1) и плохие результаты прогноза (таблицы 4, 5, дополнительная таблица 2).
Очевидное и серьезное ограничение нашего исследования заключается в небольшом количестве экспериментально аннотированных и, что наиболее важно, видоспецифичных аннотаций вторичных путей метаболизма и связанных с ними генов, что объясняется экспериментальными проблемами для определения путей и вовлеченных генов.Аннотации ферментативного пути в значительной степени основаны на переносе аннотаций на основе гомологии от модельных видов, из которых A. thaliana является наиболее значимой. Хотя было обнаружено, что A. thaliana демонстрирует более богатый, чем ожидалось, вторичный метаболизм (D’Auria and Gershenzon, 2005), использование одного или нескольких хорошо охарактеризованных видов, естественно, ограничивает возможность проверки прогностической ценности филогенетического профилирования. Важно отметить, что ограничения касаются проверяемости прогнозов.Прогнозы могут быть верными, но их невозможно сравнить с известными аннотациями. Таким образом, для дальнейшего развития и использования концепций филогенетического профилирования весьма желательным был бы расширенный набор функционально охарактеризованных и специфических генов метаболических путей у различных видов растений.
В наших анализах мы использовали только аннотированные гены ферментов. При применении филогенетического профилирования к новым геномам, конечно, не было бы известно a priori , кодирует ли ген фермент.Однако это исследование было разработано специально как технико-экономическое обоснование, чтобы можно было провести сравнение с истинными (в пределах его точности) функциональными назначениями. На практике классические методы, основанные на сравнении последовательностей, могут быть использованы в первую очередь для определения ферментативных функций, что, однако, также позволит назначать новые гены путям, если доступна соответствующая аннотация. Именно целью этого исследования было проверить, могут ли такие функциональные назначения быть сделаны на основе только сходства филогенетических профилей; я.е., не требуя подробных знаний аннотации. К сожалению, это обещание не сбылось.
Мы основали аннотацию нашей информации о метаболических путях на KEGG. Хотя KEGG пользуется большим уважением, были разработаны альтернативные базы данных по видам растений (Grafahrend-Belau et al., 2012), среди которых Сеть метаболизма растений (PMN, также известная как PlantCyc, www.plantcyc.org) представляет собой еще одно крупномасштабное растение — ресурс данных, ориентированный на метаболизм. Для сравнения мы также выполнили анализ обогащения филогенетического профиля с использованием данных PlantCyc (дополнительная таблица 3).В то время как для анализа был доступен более широкий набор из 241 пути, только пять оказались значимыми в отношении повышенного сходства филогенетических профилей. В соответствии с результатами KEGG наиболее значимым оказался путь «цикл Кальвина-Бенсона-Бассема» (скорректированный Бенджамини-Хохберг, p BH <0,001) (по данным KEGG он занял пятое место и обозначен как «Углеродный»). фиксация в фотосинтезирующих организмах »с последующей« инактивацией брассиностероидов »(p BH <0.001), «биосинтез оризалида A» (p BH = 0,048), «биосинтез 5-аминоимидазол-рибонуклеотида II» (p BH = 0,048 и «биосинтез L-аргинина II (ацетильный цикл)», p 166 BH = 0,048). Таким образом, более подробное описание пути, доступное в PlantCyc, не привело к усилению ассоциаций, хотя необходимо учитывать, что большие размеры набора (241 против 94 в KEGG) вызывают более выраженный эффект коррекции множественного тестирования. Тем не менее, мы пришли к выводу, что представленные здесь результаты не относятся к KEGG, а указывают на общую слабость этого подхода и доступность текущих данных.
Понятие об отдельных метаболических путях как подходящая абстракция функциональных взаимодействий между ферментами
В первую очередь, мы основывали функциональные ассоциации объектов генов на их вхождении в один и тот же класс путей KEGG или подробную карту путей. Таким образом, мы рассматривали пути как изолированные контейнеры, в которых все гены проявляют функциональную взаимосвязь независимо от фактического количества шагов реакции между ними. Используя это определение, были получены в основном плохие результаты статистического прогнозирования.Напротив, при изучении прямого и физического взаимодействия между ферментами филогенетические профили оказались очень информативными (рис. 10). Поскольку часто физические взаимодействия указывают на взаимосвязь между немедленными метаболическими реакциями (Durek and Walther, 2008), этот результат можно рассматривать как положительный результат исследования, указывающий также на важность расстояния метаболических путей между ферментами. Следовательно, сходство филогенетических профилей можно рассматривать как подходящую фильтрацию для идентификации истинных белок-белковых взаимодействий в экспериментальных или прогнозируемых наборах взаимодействий.По-видимому, с увеличением расстояния метаболических путей прогностическая ценность филогенетических профилей быстро снижается. В заключение, переход от контейнеров путей к сетевому расстоянию между ферментными генами кажется правильным. Это также решило бы другое очевидное ограничение контейнеров пути. Они рассматривают метаболические отношения как изолированные суб-пути, так что гены либо вовлечены в один и тот же процесс, либо вообще не связаны. Это также проиллюстрировано на рисунке 3 для пути биосинтеза дитерпеноидов.В то время как некоторые гены находятся в контейнере этого пути (карта) или встречаются у многих видов, ответвительные реакции имеют узкий видовой паттерн. Одинаковое рассмотрение всех генов на этой карте кажется неправильным и приведет к неверным выводам. Очевидно, что показатель расстояния, который отражает истинное расстояние метаболического пути, например кратчайшие пути (Durek and Walther, 2008; Walther et al., 2010), был бы предпочтительнее. Однако, несмотря на эти серьезные ограничения, мы по-прежнему рассматриваем подход, представленный в этом исследовании, как действительный первый шаг к достижению цели правильного предсказания функциональной ассоциации генов ферментов.
Выводы
В заключение, сходство филогенетических профилей оказалось нечувствительным для надежного предсказания ассоциаций генов на уровне классов и карт метаболических путей, но оказалось информативным в отношении физических взаимодействий кодируемых ферментных белков. Это исследование подчеркивает необходимость расширения наших экспериментальных знаний о путях вторичного метаболизма у разных видов растений, прежде чем можно будет сделать окончательное суждение о применимости филогенетических профилей.Это также критически отражает концепцию определения генов как функционально связанных только через членство в путях. Вместо этого представляется желательной метрика расстояния на основе сети. Положительная корреляция профилей с физическими взаимодействиями открывает возможность использовать филогенетическое профилирование в качестве этапа фильтрации для выявления истинных межбелковых взаимодействий из наборов взаимодействий-кандидатов.
Авторские взносы
DW задумал исследование, SW и DW разработали исследование, интерпретировали результаты и написали рукопись.Все вычисления были выполнены с помощью SW, за исключением прогнозов Clus-HMC и анализов коэкспрессии генов, выполняемых DW.
Заявление о конфликте интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могут быть истолкованы как потенциальный конфликт интересов.
Благодарности
Мы хотели бы поблагодарить Селин Венс, Драги Ковева и Сасо Дзероски за полезные комментарии по поводу использования программного обеспечения CLUS и интерпретации его результатов.Кроме того, мы благодарны Себастьяну Проосту, Алисдеру Ферни и Такаюки Тоге за плодотворное обсуждение проекта. Мы благодарим Кристофа Тиме за полезные комментарии к рукописи.
Дополнительные материалы
Дополнительные материалы к этой статье можно найти в Интернете по адресу: https://www.frontiersin.org/articles/10.3389/fpls.2017.01831/full#supplementary-material
Список литературы
Ольха, А., Джамиль, М., Марзорати, М., Бруно, М., Верматен, М., Bigler, P., et al. (2012). Путь от бета-каротина к карлактону, стриголактоноподобному гормону растений. Science 335, 1348–1351. DOI: 10.1126 / science.1218094
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ашкенази, С., Снир, Р., Офран, Ю. (2012). Оценка взаимосвязи между сохранением функции и сохранением последовательности с использованием фотосинтетических белков. Биоинформатика 28, 3203–3210. DOI: 10.1093 / биоинформатика / bts608
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ченг, Ю., и Perocchi, F. (2015). ProtPhylo: идентификация белков-фенотипов и функциональных ассоциаций белок-белок с помощью филогенетического профилирования. Nucleic Acids Res 43, W160 – W168. DOI: 10.1093 / nar / gkv455
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Чу, Х.Й., Вегель, Э., Осборн, А. (2011). От гормонов к вторичному метаболизму: появление метаболических кластеров генов у растений. Plant J. 66, 66–79. DOI: 10.1111 / j.1365-313X.2011.04503.x
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Крейгон, Д. Дж., Джеймс, Н., Окайере, Дж., Хиггинс, Дж., Джотэм, Дж. И Мэй, С. (2004). NASCArrays: хранилище данных микрочипов, созданных службой транскриптомики NASC. Nucleic Acids Res 32: D575. DOI: 10.1093 / nar / gkh233
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Csardi, G., and Nepusz, T. (2006). Программный пакет Igraph для комплексного исследования сети .Межжурнальные сложные системы.
Google Scholar
Д’Аурия, Дж. К., и Гершензон, Дж. (2005). Вторичный метаболизм Arabidopsis thaliana : растет как сорняк. Curr. Opin. Plant Biol. 8, 308–316. DOI: 10.1016 / j.pbi.2005.03.012.
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Дэвис Дж. И Гоадрич М. (2006). «Взаимосвязь между прецизионным воспроизведением и кривыми ROC», Труды 23-й Международной конференции по машинному обучению (Питтсбург, Пенсильвания), 233–240.
Google Scholar
Дурек П. и Вальтер Д. (2008). Интегрированный анализ сетей метаболического и белкового взаимодействия раскрывает новые принципы организации молекул. BMC Syst. Биол. 2: 100. DOI: 10.1186 / 1752-0509-2-100
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Эммс, Д. М., и Келли, С. (2015). OrthoFinder: устранение фундаментальных ошибок при сравнении всего генома значительно повышает точность вывода ортогруппы. Genome Biol 16: 157. DOI: 10.1186 / S13059-015-0721-2
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Фрейли К. и Рафтери А. Э. (1999). MCLUST: программное обеспечение для кластерного анализа на основе моделей. J. Classific. 16, 297–306. DOI: 10.1007 / s003579
8CrossRef Полный текст | Google Scholar
Gaasterland, T., and Ragan, M.A. (1998). Микробные гены: филетические и функциональные паттерны распределения ORF среди прокариот. Microb. Комп. Геномика 3, 199–217. DOI: 10.1089 / omi.1.1998.3.199
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Гачон, К. М., Ланглуа-Мёринн, М., Генри, Ю., и Сайндренан, П. (2005). Совместная транскрипционная регуляция ферментов вторичного метаболизма арабидопсиса: функциональные и эволюционные последствия. Plant Mol. Биол. 58, 229–245. DOI: 10.1007 / s11103-005-5346-5
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Grafahrend-Belau, E., Юнкер, Б. Х., и Шрайбер, Ф. (2012). «Метаболические пути растений: базы данных и конвейер для стехиометрического анализа», в Seed Development: OMICS Technologies к улучшению качества семян и урожайности (Dordrecht: Springer), 345–366.
Google Scholar
Хартманн, Т. (1996). Разнообразие и изменчивость вторичного метаболизма растений: механистический взгляд. Энтомол. Exper. Et Appl. 80, 177–188.
Google Scholar
Джоти, Р., Пржитицка, Т. М., и Аравинд, Л. (2007). Обнаружение функциональных связей и не охарактеризованных клеточных путей с использованием сравнений филогенетических профилей: всесторонняя оценка. BMC Bioinformat. 8: 173. DOI: 10.1186 / 1471-2105-8-173
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Керси, П. Дж., Аллен, Дж. Э., Армеан, И., Бодду, С., Болт, Б. Дж., Карвалью-Силва, Д. и др. (2016). Ensembl Genomes 2016: больше геномов, больше сложности. Nucleic Acids Res. 44, D574 – D580. DOI: 10.1093 / nar / gkv1209
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Херсонский О., Рудвельдт К., Тауфик Д. С. (2006). Ферментная неразборчивость: эволюционные и механистические аспекты. Curr. Opin. Chem. Биол. 10, 498–508. DOI: 10.1016 / j.cbpa.2006.08.011
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ким, Ю., и Субраманиам, С. (2006). Локально определенные филогенетические профили белков выявляют ранее пропущенные белковые взаимодействия и функциональные взаимосвязи. Белки 62, 1115–1124. DOI: 10.1002 / prot.20830
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Kinsella, R.J., Kähäri, A., Haider, S., Zamora, J., Proctor, G., Spudich, G., et al. (2011). Ensembl BioMarts: центр поиска данных в таксономическом пространстве. База данных 2011: bar030. DOI: 10.1093 / база данных / bar030
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Коркуч П., Шипперс Дж. Х. и Вальтер Д.(2014). Характеристика и идентификация цис-регуляторных элементов у Arabidopsis на основе информации о однонуклеотидном полиморфизме. Plant Physiol. 164, 181–200. DOI: 10.1104 / стр.113.229716
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ло Конте, Л., Бреннер, С. Э., Хаббард, Т. Дж., Чотиа, К., и Мурзин, А. Г. (2002). База данных SCOP в 2002 году: уточнения учитывают структурную геномику. Nucleic Acids Res. 30, 264–267. DOI: 10.1093 / нар / 30.1.264
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Lohse, M., Nagel, A., Herter, T., May, P., Schroda, M., Zrenner, R., et al. (2014). Mercator: быстрый и простой веб-сервер для функциональной аннотации данных последовательностей растений в масштабе генома. Plant Cell Environ. 37, 1250–1258. DOI: 10.1111 / pce.12231
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Оксман-Кальдентей, К. М., и Инзе, Д. (2004). Фабрики растительных клеток в постгеномную эру: новые способы производства вторичных метаболитов. Trends Plant Sci . 9, 433–440. DOI: 10.1016 / j.tplants.2004.07.006
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Оренго, К. А., Брей, Дж. Э., Бьюкен, Д. У., Харрисон, А., Ли, Д., Перл, Ф. М. и др. (2002). База данных семейства белков CATH: ресурс для структурной и функциональной аннотации геномов. Proteomics 2, 11–21. DOI: 10.1002 / 1615-9861 (200201) 2: 1 <11 :: AID-PROT11> 3.0.CO; 2-T
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Пеллегрини, М., Маркотт, Э. М., Томпсон, М. Дж., Айзенберг, Д., и Йейтс, Т. О. (1999). Назначение функций белков с помощью сравнительного анализа генома: филогенетические профили белков. Proc. Natl. Акад. Sci. США 96, 4285–4288.
PubMed Аннотация | Google Scholar
Пичерский, Э., и Банда, Д. Р. (2000). Генетика и биохимия вторичных метаболитов растений: эволюционная перспектива. Trends Plant Sci. 5, 439–445. DOI: 10.1016 / S1360-1385 (00) 01741-6
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ранея, Дж.А., Йейтс, К., Грант, А., и Оренго, К. А. (2007). Прогнозирование функции белков с помощью иерархических филогенетических профилей: метод Gene3D Phylo-Tuner, применяемый к эукариотическим геномам. PLoS Comput. Биол. 3: e237. DOI: 10.1371 / journal.pcbi.0030237
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Сандер К. и Шнайдер Р. (1991). База данных гомологичных белковых структур и структурное значение выравнивания последовательностей. Белки 9, 56–68.DOI: 10.1002 / prot.3400
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Шауэр Н., Семел Ю., Ресснер У., Гур А., Бальбо И., Каррари Ф. и др. (2006). Комплексное метаболическое профилирование и фенотипирование линий межвидовой интрогрессии для улучшения томатов. Nat. Biotechnol. 24, 447–454. DOI: 10.1038 / nbt1192
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Schietgat, L., Vens, C., Struyf, J., Blockeel, H., Кочев, Д., Дзероски, С. (2010). Прогнозирование функции генов с использованием иерархических ансамблей дерева решений с несколькими метками. BMC Bioinformatics 11: 2. DOI: 10.1186 / 1471-2105-11-2
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Шмидт Б. М., Рыбницки Д. М., Липски П. Э. и Раскин И. (2007). Возвращаясь к древней концепции ботанической терапии. Nat. Chem. Биол. 3, 360–366. DOI: 10.1038 / nchembio0707-360
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Сингх, Б., и Бхат, Т. К. (2003). Возможные терапевтические применения некоторых вторичных метаболитов растений, являющихся антипитательными веществами. J. Agric. Food Chem. 51, 5579–5597. DOI: 10.1021 / jf021150r
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Skunca, N., Bošnjak, M., Kriško, A., Panov, P., Džeroski, S., Smuc, T., et al. (2013). Филетическое профилирование кликами ортологов усиливается признаками паралогических отношений. PLoS Comput. Биол. 9: e1002852.DOI: 10.1371 / journal.pcbi.1002852
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Ternes, P., Sperling, P., Albrecht, S., Franke, S., Cregg, J. M., Warnecke, D., et al. (2006). Идентификация грибковых сфинголипидных C9-метилтрансфераз с помощью филогенетического профилирования. J. Biol. Chem. 281, 5582–5592. DOI: 10.1074 / jbc.M512864200
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Вилелла, А. Дж., Северин, Дж., Урета-Видаль, А., Хенг, Л., Дурбин, Р., и Бирни, Э. (2009). EnsemblCompara GeneTrees: полные филогенетические деревья позвоночных с учетом дублирования. Genome Res. 19, 327–335. DOI: 10.1101 / gr.073585.107
PubMed Аннотация | CrossRef Полный текст | Google Scholar
Вальтер, Д., Страсбург, К., Дурек, П., и Копка, Дж. (2010). Взаимосвязи метаболических путей, выявленные интегративным анализом транскрипционной и метаболической динамики температурного стресс-ответа у дрожжей. OMICS 29, 944–959.DOI: 10.1089 / omi.2010.0010
CrossRef Полный текст | Google Scholar
Wisecaver, J. H., Borowsky, A. T., Tzin, V., Jander, G., Kliebenstein, D. J., and Rokas, A. (2016). Подход глобальной сети коэкспрессии для подключения генов к специализированным метаболическим путям у растений. bioRxiv . 29, 944–959. DOI: 10.1101 / 093914
CrossRef Полный текст | Google Scholar
Йенчо, Г. К., Ковальски, С. П., Кобаяши, Р. С., Синден, С. Л., Боньербале, М.В., и Деаль, К. Л. (1998). QTL-картирование гликоалкалоидных агликонов листвы в потомстве картофеля Solanum tuberosum x S-berthaultii : количественные вариации и вторичный метаболизм растений. Теор. Прил. Genet. 97, 563–574. DOI: 10.1007 / s001220050932
CrossRef Полный текст | Google Scholar
Техника исследования возможных метаболических путей с использованием скрытого химического пространства | Биоинформатика
Абстрактные
Мотивация
Изучение метаболических путей — один из ключевых методов разработки высокопродуктивных микробов для биопродукции химических соединений.Для изучения возможных путей необходимы не только изучение комбинации хорошо известных ферментативных реакций, но и поиск потенциальных ферментативных реакций, которые могут катализировать желаемые структурные изменения. Для достижения этого в большинстве традиционных методик используются правила заранее заданных вручную реакций, однако они не могут в достаточной степени обнаружить потенциальные реакции, поскольку обычные правила не могут полностью выразить структурные изменения до и после ферментативных реакций. Оценка осуществимости изученных путей является еще одной проблемой, потому что нет способа подтвердить возможность реакции неизвестных ферментативных реакций с помощью этих правил.Таким образом, методика комплексной регистрации структурных изменений ферментативных реакций и методика оценки осуществимости пути по-прежнему необходимы для изучения возможных путей метаболизма.
Результаты
Мы разработали технику исследования возможных путей с использованием скрытого химического пространства, полученного из глубинной генеративной модели для сложных структур. С помощью этого метода ферментативная реакция рассматривается как вектор различия между основным субстратом и основным продуктом в химическом латентном пространстве, полученном из генеративной модели.Особенности ферментативной реакции встроены в вектор с фиксированной размерностью, и можно всесторонне выразить структурные изменения ферментативных реакций. Этот метод также включает выбор реакции на основе дифференциальной эволюции для разработки возможных возможных путей и оценку путей с использованием прогнозирования возможности реакции на основе нейронной сети. Предложенный метод был применен к незарегистрированным путям, имеющим отношение к производству 2-бутанона, и успешно исследованы возможные пути, которые включают такие реакции.
1 Введение
Микробиологическое производство химических соединений вносит важный вклад в развитие устойчивых производств. Поскольку создание высокопродуктивного микроба часто требует огромного количества времени и усилий, технологии проектирования и построения биологических функций микробов на компьютерах становятся все более важными для сокращения периода развития таких микробов. Важным этапом в технологии микробного дизайна in silico является разработка метаболического пути, в котором определяется серия ферментативных реакций, которые способствуют желаемым химическим структурным изменениям от исходного соединения (метаболита) до целевого соединения (Choi et al., 2019). Помимо выбора промежуточных соединений, среди промежуточных соединений должны быть найдены потенциальные ферменты, катализирующие химические реакции. А именно, чтобы разработать высокопроизводительные метаболические пути, необходимо не только изучить комбинацию хорошо известных ферментативных реакций, но и найти комбинацию потенциальных ферментативных реакций, которые могут катализировать желаемые структурные изменения. Поскольку для изучения всех возможных метаболических путей, которые включают такие потенциальные реакции, требуется много ручных и вычислительных усилий, все еще необходим эффективный метод исследования таких путей, чтобы сократить время развития высокопродуктивных микробов.
Хотя были предложены различные методы исследования метаболических путей in silico (Wang et al. , 2017), остаются три основных технических проблемы для эффективного исследования метаболических путей; (i) как представить ферментативную реакцию в компьютерной системе, (ii) как разработать возможные пути-кандидаты, комбинируя огромное количество потенциальных ферментативных реакций, и (iii) как оценить актуальность возможных путей-кандидатов. Для решения первой задачи в большинстве традиционных методов используется метод представления реакции, который включает в себя ручную подготовку заранее определенных правил реакции и определение изменений в субструктуре, фокусирующейся вблизи центра реакции, на основе правил реакции (Araki et al., 2014 г .; Делепин и др. , 2018; Хадади и Хатзиманикатис, 2015; Kumar et al. , 2018; Мория и др. , 2010 г.). Хотя этот метод представления точно идентифицирует небольшие изменения в частичных структурах, таких как функциональная группа, они недостаточно идентифицируют общие структуры основы, участвующие в субстратной специфичности ферментативных реакций. Для решения второй задачи традиционные методы часто исследуют возможные пути с помощью логических операций на основе правил, таких как добавление и удаление функциональных групп или атомов.Их преимущество в том, что нереалистичные пути, включающие нереалистичные составные структуры и ферментативные реакции, не исследуются. Однако возможные пути развития недостаточно изучены, поскольку эти методы не учитывают ферментативные реакции, отсутствующие в рабочем правиле. Для третьей задачи проверка актуальности неизвестных ферментативных реакций также становится проблемой с традиционными методами, основанными на правилах. Следовательно, необходимо химическое внедрение, которое может количественно определять особенности составных структур более точно, чем традиционные методы, такие как вариационный автокодировщик (VAE).
Мы предлагаем метод исследования возможных метаболических путей с использованием скрытого химического пространства, полученного из глубинной генеративной модели для сложных структур. Недавно была предложена глубокая генеративная модель для соединений для сопоставления структуры соединения, описанной в стилях упрощенной молекулярной системы ввода линии ввода (SMILES), в латентное векторное пространство (Gómez-Bombarelli et al. , 2018; Jin et al. , 2018; Куснер и др. , 2017). Используя химическое латентное пространство, этот метод включает в себя метод, с помощью которого ферментативные реакции представлены как вектор различия между латентными векторами основного субстрата и основного продукта.Используя представление метаболической реакции, можно не только определять изменения в общей структуре скелета, связанные со специфичностью субстрата, но также устранять необходимость в правилах реакции, которые требовались для каждой реакции, чтобы реакции могли выполняться единообразно. Благодаря идентичным размерам латентных векторов, латентный вектор (-ы) промежуточного соединения (-ов) между исходным и целевым соединениями может быть выражен простой математической операцией среди векторов признаков реакции.Более того, латентный вектор каждого промежуточного соединения может быть реконструирован с использованием глубинной генеративной модели и использован для новых составных структур. Мы также разработали методику проектирования возможных путей-кандидатов на основе дифференциальной эволюции (DE) путем комбинирования потенциальных ферментативных реакций и метод прогнозирования возможности реакции на основе нейронной сети (NN) для оценки релевантности потенциальных ферментативных реакций и возможные пути в качестве возможных путей. Этот метод проектирования выбирает вектор (ы) признаков реакции и минимизирует квадратичную ошибку между вектором признаков пути, который был вычислен латентными векторами исходного и целевого соединений, и суммой выбранных векторов признаков реакции.Метод подсчета очков вычисляет значение возможности реакции и общий балл пути каждой реакции с учетом специфичности субстрата в латентном пространстве. Чтобы проверить эффективность предложенного метода, мы применили его к задачам исследования путей, которые включают как зарегистрированные, так и незарегистрированные реакции.
2 Материалы и методы
2.1 Исследование возможных путей метаболизма и связанные с ними работы
Исследование метаболических путей включает обнаружение серии ферментативных реакций, которые способствуют желаемому структурному изменению химических веществ от исходного соединения до целевого соединения.Во время исследования необходимо не только изучить все пути, состоящие из нескольких известных ферментативных реакций, зарегистрированных в курируемых базах данных биологических путей (БД), таких как Киотская энциклопедия генов и геномов (KEGG) (Kanehisa and Goto, 2000), Metacyc (Caspi и др. , 2018) и MetaNetX (Moretti и др. , 2016), но также возможные пути, состоящие из ферментативных реакций, которые не были зарегистрированы в БД, но потенциально катализируют промежуточные соединения (рис.1).
Рис. 1.
Исследование возможного метаболического пути. Часто существует несколько путей получения целевого соединения из исходного соединения в метаболической системе. В дополнение к таким известным путям могут быть включены неизвестные ферментативные реакции и соединения, не зарегистрированные в базе данных (БД). А именно, может существовать несколько возможных путей от исходного соединения к целевому соединению, которые включают как зарегистрированные (серые сплошные линии), так и незарегистрированные (оранжевые пунктирные линии) реакции
Рис.1.
Исследование возможных путей метаболизма. Часто существует несколько путей получения целевого соединения из исходного соединения в метаболической системе. В дополнение к таким известным путям могут быть включены неизвестные ферментативные реакции и соединения, не зарегистрированные в базе данных (БД). А именно, может существовать несколько возможных путей от исходного соединения к целевому соединению, которые включают как зарегистрированные (серые сплошные линии), так и незарегистрированные (оранжевые пунктирные линии) реакции.
Для эффективного исследования таких возможных путей в компьютерных системах полезно: использовать метод, при котором каждая ферментативная реакция рассматривается как арифметическое выражение, т.е.е. векторы или матрицы, и весь путь может быть вычислен как арифметическая суперпозиция отдельных реакций. Используя такой математический метод, структурное изменение между субстратом и продуктом, катализируемое ферментативной реакцией, можно выразить как добавление, замещение или перегруппировку; таким образом, можно легко создать и изучить множество потенциальных ферментативных реакций и промежуточных соединений с помощью вычислительных алгоритмов.
Были предложены представления реакций, такие как молекулярные отпечатки пальцев, основанные на определенных методах подсчета субструктур, которые представляют составную структуру с вектором, состоящим из числа определенных частичных структур, составляющих всю структуру (Araki et al., 2014 г .; Kumar et al. , 2018). Они также определяют вектор различия, полученный путем вычитания вектора структурных признаков основного субстрата из основного продукта в качестве признака реакции и связанный с номером комиссии по ферментам (EC), используя информацию о метаболических путях KEGG. Следовательно, можно получить векторы структурных признаков соединения-продукта путем добавления произвольного вектора признаков реакции к векторам структурных признаков соединения-субстрата.
Одна проблема в таком традиционном векторном представлении на основе отпечатков пальцев состоит в том, что вектор молекулярных отпечатков пальцев не может воспроизводить составную структуру, потому что он не имеет информации о связности между частичными структурами. В случае «отпечатка пальца» неизвестного соединения, катализируемого потенциальными ферментами, можно указать известную аналогичную структуру соединения, только выполнив поиск структуры. Кроме того, в случае соединений, имеющих разные абсолютные конфигурации, т.е.е. изомеры, даже известные соединения не могут быть различимы. Для решения этой проблемы необходимо разработать метод исследования пути с использованием другого математического метода ферментативных реакций, который удовлетворяет следующим двум пунктам:
Соединения метаболического пути могут быть выражены в распределенном представлении с пространством признаков фиксированный размер.
Вектор структурных признаков продукта после добавления вектора признаков реакции к вектору структурных признаков подложки может быть декодирован в составную структуру без потери информации о связности.
Глубокая генеративная модель химических соединений, известная как молекулярный автоэнкодер, представляет собой инновационный метод составной экспрессии, основанный на вариационном байесовском методе, в котором строки SMILES соединений кодируются в фиксированном измерении латентных векторов ( Гомес-Бомбарелли и др. , 2018). Поэтому для удовлетворения вышеуказанных требований в предлагаемой технике используются латентные векторы, основанные на дереве соединений VAE (JT-VAE), которое является современной глубокой генеративной моделью для химических соединений (Jin et al., 2018).
2.2 Предлагаемый метод разведки возможных путей
2.2.1 Общая структура
Рисунок 2 иллюстрирует общую структуру предлагаемого метода. Техника условно разделена на два этапа: вычисление характеристик реакции и исследование пути. На первом этапе характеристики реакций соединений на метаболическом пути вычисляются как векторы признаков с использованием глубокой генеративной модели и накапливаются в базе данных признаков реакции.Исследование пути состоит из выбора признаков-реакций, в котором пути-кандидаты исследуются с использованием векторов признаков, хранящихся в базе данных признаков-реакций, и оценки путей, в которой наиболее релевантный путь выбирается из путей-кандидатов.
Рис. 2.
Обзор предлагаемой методики. Он включает в себя этапы вычисления характеристик реакции и исследования пути. При вычислении характеристик реакции модели вариационного автоэнкодера (VAE) обучаются с помощью общедоступной составной БД.Затем с использованием латентных векторов соединений вычисляются векторы признаков реакции. Исследование пути состоит из дизайна пути и оценки пути. А именно, разработаны и оценены несколько возможных путей.
Рис. 2.
Обзор предлагаемой техники. Он включает в себя этапы вычисления характеристик реакции и исследования пути. При вычислении характеристик реакции модели вариационного автоэнкодера (VAE) обучаются с помощью общедоступной составной БД. Затем с использованием латентных векторов соединений вычисляются векторы признаков реакции.Исследование пути состоит из дизайна пути и оценки пути. А именно, разработаны и оценены несколько возможных путей
2.2.2 Векторы признаков реакции с использованием химического латентного пространства
Как упоминалось выше, мы используем кодировщики на основе JT-VAE для кодирования определенной структуры химического соединения в скрытый вектор. На рисунке 3 показан обзор JT-VAE. Он имеет два типа кодировщиков. Один из них — кодировщик графа, а другой — кодировщик дерева. Разложение дерева на основе метода дерева признаков (Rarey and Dixon, 1998) проводится для оценки молекулярного сходства между небольшими органическими соединениями.Вместо линейного представления, такого как отпечатки пальцев, для молекулы рассчитывается более сложное описание, дерево характеристик. Такая характеристика дерева соединений эффективна для представления общей структуры скелета соединений; таким образом, ожидается, что древовидный латентный вектор, кодируемый с помощью JT-VAE, также будет представлять составную структуру.
Рис. 3.
Архитектура дерева соединений VAE (JT-VAE) (Jin et al. , 2018). JT-VAE имеет два кодировщика, граф и дерево.Входной сигнал древовидного кодировщика представляет собой дерево соединений, разложенное с использованием техники дерева признаков (Rarey and Dixon, 1998). Цветной узел в дереве признаков представляет подструктуру соединения
Рис. 3.
Архитектура дерева соединений VAE (JT-VAE) (Jin et al. , 2018). JT-VAE имеет два кодировщика, граф и дерево. Входной сигнал древовидного кодировщика представляет собой дерево соединений, разложенное с использованием техники дерева признаков (Rarey and Dixon, 1998). Цветовой узел в дереве признаков представляет подструктуру соединения
. На рисунке 4 показан метод генерации векторов признаков реакции с использованием кодеров JT-VAE.Сначала кодировщики обучаются с использованием составного набора данных в составной БД перед вычислением векторов характеристик реакции. Затем информация о метаболических путях, такая как «гликолиз», анализируется из базы данных метаболических путей, таких как KEGG. Затем строка SMILES соединения метаболического пути вводится обученным кодировщикам JT-VAE. Генерируется скрытый вектор (zC00267) соединения и отображается на скрытое пространство размером N . Вектор признаков реакции генерируется путем вычитания латентного вектора основного субстрата из вектора основного продукта с помощью следующего уравнения: где zpro — латентный вектор соединения-продукта, а zsub — это вектор субстратного соединения, как показано на рисунке. 4.Рис. 4.
Объяснение вектора-признака реакции. Во-первых, латентные векторы соединений, зарегистрированных в БД метаболического пути, получают от кодировщиков JT-VAE. Затем, используя латентные векторы основного субстрата и продукта на основе DBs метаболических путей, получают вектор признаков реакции, который определяется как вектор различия этих латентных векторов. Вектор характеристики реакции EC2.7.1.1 вычитает гидроксигруппу и добавляет фосфатную группу к α -d-глюкозе
Рис.4.
Объяснение вектора-признака реакции. Во-первых, латентные векторы соединений, зарегистрированных в БД метаболического пути, получают от кодировщиков JT-VAE. Затем, используя латентные векторы основного субстрата и продукта на основе DBs метаболических путей, получают вектор признаков реакции, который определяется как вектор различия этих латентных векторов. Вектор признаков реакции EC2.7.1.1 вычитает гидроксигруппу и добавляет фосфатную группу к α -d-глюкоза
Таким образом, все реакции метаболических путей в БД метаболических путей кодируются векторами признаков реакции и хранится в БД признаков реакции.Одновременно записывается каждый вектор признаков реакции, и ему присваивается номер EC. На рисунке 4, например, вектор-признак реакции rec2.7.1.1, сгенерированный из латентных векторов zC00267 и zC00668 основного субстрата α -d-глюкоза (идентификатор соединения KEGG: C00267) и основного продукта α -d-глюкозо-6-фосфат ( Код соединения KEGG: C00668) зарегистрирован с номером EC 2.7.1.1.
2.2.3 Разработка возможных путей развития
На рис. 5 показана процедура этапа проектирования возможных путей.Этот шаг состоит из (i) вычисления характеристик пути, (ii) генерации случайного подмножества, (iii) выбора реакции, (iv) упорядочения комбинационных реакций и (v) удаления нереалистичных путей.
Рис. 5.
Процедура проектирования возможных путей. Наборы векторов признаков-реакций выбираются с использованием метода оптимизации для минимизации квадратичной ошибки между вектором признаков-путей и суммой выбранных векторов признаков-реакций. На этом рисунке показан пример, в котором выбраны три вектора признаков реакции (ra, rb и rc).Всего существует шесть комбинационных порядков. Промежуточные соединения реконструируются с использованием декодера JT-VAE. Наконец, нереалистичный путь (пути) удаляется на основании изменений молекулярной массы, а оставшиеся пути-кандидаты добавляются в список путей-кандидатов
Рис. 5.
Процедура проектирования путей для путей-кандидатов. Наборы векторов признаков-реакций выбираются с использованием метода оптимизации для минимизации квадратичной ошибки между вектором признаков-путей и суммой выбранных векторов признаков-реакций.На этом рисунке показан пример, в котором выбраны три вектора признаков реакции (ra, rb и rc). Всего существует шесть комбинационных порядков. Промежуточные соединения реконструируются с использованием декодера JT-VAE. Наконец, нереалистичные пути удаляются на основании изменений молекулярной массы, а оставшиеся пути-кандидаты добавляются в список
путей-кандидатов. целевое соединение ztarget из начального соединения zstart (рис.5 (1)). А именно, вектор p признаков пути выводится из следующего уравнения:Затем векторы признаков реакции M случайным образом выбираются из базы данных признаков реакции. Выбранные векторы определяются как подмножество реакций, чтобы уменьшить объем вычислений и повысить эффективность поиска (рис. 5 (2)).
Затем набор векторов признаков реакции для проектирования путей определяется с использованием метода оптимизации, чтобы минимизировать квадратичную ошибку между вектором признаков пути и суммой векторов признаков реакции в наборе (рис.5 (3)). Другими словами, целевая функция оптимизации определяется следующим уравнением:min | e | 2 = ∑j = 0N-1pj-∑i = 0M-1xiri, j2s.t. | E | ≤Th, ∑ i = 1Mxi≤K, xj∈ {0,1,2, …, K}
(3) где p j — значение вектора пути-признака p из j -го измерения , x i — целочисленное значение для i -го индекса подмножества, ri, j — значение вектора признаков реакции для i -го индекса подмножества и j -го измерения, Th — порог ошибки и K — максимальное количество стадий реакции.Поскольку целевая функция использует квадратную ошибку e между суммой выбранных векторов признаков реакции и вектора признаков пути, предусмотрена функция штрафа, которая нелинейно увеличивается в соответствии с максимальным числом шагов реакции. А именно, проблема минимизации рассматривается как проблема нелинейного целочисленного программирования (НЛП).
Чтобы решить эту проблему НЛП, мы применяем технику DE (Storn and Price, 1997) из-за ее высокой производительности поиска, несмотря на то, что это простой алгоритм.Мы вводим функцию пригодности f (x) в метод DE, которая получается из квадратичной ошибки | e | 2 среди векторов признаков и штрафной функции λ , которая нелинейно увеличивается в соответствии с максимальным числом предусмотренных шагов реакции. как условие ограничения.мин f (x) = | e | 2 + λ → min
(4)λ = C × exp (xlen) 2 (xlen> K) 0,0 (xlen≤K)
(5) где x len — количество выбранных реакций, а C — постоянный параметр. Каждый человек в исходной популяции инициализируется таким образом, чтобы сумма элементов находилась в пределах максимального количества шагов реакции.Используется двоичный алгоритм DE, в котором значение каждого элемента после вычисления эволюции округляется для решения проблемы НЛП. Бинарный алгоритм DE для выбора признаков реакции описывается как алгоритм 1. Применяя двоичный алгоритм DE к выбору признаков реакции, получается набор векторов признаков реакции в виде набора векторов компонентов для разработки желаемого вектора признаков пути. .Algorithm 1Алгоритм двоичного DE для выбора реакции
Исходная популяция P ( g = 0) составляет P случайным образом сгенерировано особей
Оценить P ( g = 0)
Установить длину индивидуума как размер подмножества реакций M
для поколения g = от 1 до Окончание do
для индивидуально p = 0 до 12 do0007 — 1 — 1 Сгенерировать случайные числа a , b , c ∈ [0, P — 1]
Выберите трех отдельных лиц xa, xb, xc в качестве родители
для параметра j = от 0 до M -1 do
Вычислить мутатор vj = xa, j + F (xb, j − xc, j)
Изменить значение на двоичный 0 или 1 следующим образом:
, если vj> = 0.5 , затем
Установить vj = 1,0
else
Установить vj = 0,0
end if
Вычислить кроссовер следующим образом:
Создать равномерный случайный rnd≡U (0,1)
if rnd < CR then
Установить u k = v j
else
Установить uk = xp12 902
end
конец для
если f (u)
Замените xp на u
конец, если
конец для
конец для всех Затем конструируются пути путем упорядочивания всех комбинаций векторов признаков реакции в наборе (рис.5 (4)). Одновременно с этим реконструируются промежуточные соединения в возможных путях с использованием декодера JT-VAE. А именно, векторы признаков реакции последовательно добавляют к латентному вектору исходного соединения, получая при этом латентные векторы промежуточных соединений в каждом сегменте путей-кандидатов. Эти скрытые векторы промежуточных соединений затем воспроизводятся декодером JT-VAE в виде строки SMILES со сложной структурой.
Наконец, мы оцениваем возможные пути и удаляем нереалистичные следующим образом (рис.5 (5)). В процессе упорядочивания векторов признаков реакции на рисунке 5 (4) промежуточные соединения, имеющие нереалистичную структуру, часто включаются в возможные пути из-за неоднозначных характеристик скрытого пространства JT-VAE. Чтобы исключить такой путь, мы рассчитываем изменения молекулярной массы субстрата и продукта на каждом участке пути-кандидата. А именно, мы опускаем путь, включающий сегмент, имеющий большее изменение молекулярной массы, чем предварительно определенный порог.
Повторяя шаги (i) — (v) несколько раз, можно получить пути-кандидаты от исходного соединения до целевого соединения.
2.2.4 Оценка возможных путей развития
Используя векторы реакции-признаков, мы также разработали метод оценки путей для оценки осуществимости возможных путей, разработанных в соответствии с методом, описанным в предыдущем разделе. Прогнозирование возможности реакции выполняется с использованием схемы голосования, которая усредняет выходные данные наборов дискриминаторов, обученных с различными наборами данных.
Схема голосования — это эффективный метод вывода значений прогноза с точки зрения снижения уровня отклонения и / или повышения уровня точности (Battiti and Colla, 1994). Общая бинарная классификация не может хорошо справиться с реальными реакциями, которые ошибочно считаются виртуальными, или реакциями, которые имеют возможность реакции, которая действительно имела бы место, но помечена как виртуальная реакция. «Виртуальный» означает, что реакция виртуально рассчитывается на компьютере и не регистрируется в KEGG.Чтобы решить эту проблему, мы избегаем полного отказа, используя схему голосования для вывода ансамбля NN. То есть выход не является значением реального (1) или виртуального (0) значения, а является значением возможности реакции от 0,0 до 1,0 с неоднозначностью.
Рисунок 6a иллюстрирует ансамбль NN и обучение каждой NN. Каждое значение возможности реакции v r получается из ансамбля NN. Каждый NN принимает входные данные в виде пары вектора признаков реакции и вектора скрытого субстрата и выдает 0 или 1.При обучении получается несколько весов при выполнении перекрестной проверки в R раз для каждого набора данных (количество наборов данных составляет Q ). Зарегистрированные данные ферментативной реакции устанавливаются как реальные данные (обозначены как 1.0), а незарегистрированные данные устанавливаются как виртуальные данные (обозначенные как 0.0). Общее количество моделей NN — Q × R . Берется среднее значение этих выходов, затем v r вычисляется от 0,0 до 1,0.
Рис.6.
Метод оценки возможных путей. ( a ) Ансамбль нейронных сетей (NN) используется для прогнозирования значения вероятности реакции. Множественные веса модели NN получаются в результате обучения с использованием каждого набора данных. Каждый NN выводит 0 или 1. Значение возможности реакции v r от 0,0 до 1,0 окончательно получается с использованием схемы голосования. ( b ) Это пример значения осуществимости пути v p путем умножения трех значений возможности реакции векторов признаков реакции (vr1, vr2 и vr3)
Рис.6.
Метод оценки возможных путей. ( a ) Ансамбль нейронных сетей (NN) используется для прогнозирования значения вероятности реакции. Множественные веса модели NN получаются в результате обучения с использованием каждого набора данных. Каждый NN выводит 0 или 1. Значение возможности реакции v r от 0,0 до 1,0 окончательно получается с использованием схемы голосования. ( b ) Это пример значения осуществимости пути v p путем умножения трех значений возможности реакции векторов признаков реакции (vr1, vr2 и vr3)
На рисунке 6b показано, как значение осуществимости пути v p каждого пути-кандидата получается, когда три реакции выбираются на пути от исходного соединения к целевому соединению.Когда выбраны три реакции, будет два промежуточных соединения. То есть в латентном пространстве вектор признаков пути от латентного вектора S исходного соединения до вектора целевого соединения T представлен тремя векторами признаков реакции (r1, r2 и r3). Кроме того, два промежуточных соединения представлены как Ia и Ib. В каждой реакции латентный вектор субстрата и вектор признаков реакции вводятся в вышеуказанный ансамбль NN для получения v r .Умножая все полученные v r s, получается v p .
Затем пути-кандидаты сортируются по баллу s , вычисленному с абсолютной ошибкой | e | и v p , как показано в уравнении (6).3 результат
3.1 Наборы данных и обучение VAE
Набор данных для обучения VAE SMILES состоял из набора данных ZINC (Sterling and Irwin, 2015), используемого в JT-VAE, и данных о соединениях, полученных из базы данных метаболических путей, KEGG.Строки SMILES соединений БД метаболического пути были приобретены у PubChem (Kim et al. , 2016) и ChEBI (Degtyarenko et al. , 2007). В наборе обучающих данных были исключены соединения, содержащие символ «*», обозначающий подстановочный знак, и «.» Ионной связи. В этих условиях было подготовлено более 260 тыс. Данных о соединениях. Количество эпох обучения было установлено равным 10. Применяя разложение по дереву для 260K молекул, мы собрали наш словарный набор V размером | V | = 1279.Размер скрытого состояния был установлен как 450 для всех модулей в JT-VAE, а размер скрытого узкого места был установлен как 56 со ссылкой на JT-VAE (Jin et al. , 2018).
Набор данных по ферментативным реакциям для разработки путей состоял из 9794 частей данных о реакциях, полученных из баз данных метаболических путей. Каждая часть данных включает номер ЕС и пару реакций основного субстрата и основного продукта. Используя обученный кодировщик JT-VAE, латентные векторы соединений были получены из БД метаболического пути (Kanehisa and Goto, 2000) и БД соединений (Дегтяренко et al., 2007; Kim et al. , 2016). Векторы реакций из набора данных ферментативных реакций были созданы с использованием химически-латентных векторов. Затем каждый вектор реакции был записан в набор данных и ему был присвоен номер EC.
При обучении прогнозированию возможности реакции на основе NN для оценки пути использовались четыре типа наборов данных. Подробная информация о виртуальных наборах данных приведена в Разделе 3.5.1.
3.2 Представление реакции
3.2.1 Реконструкция соединений метаболического пути
Хотя исследование JT-VAE с использованием набора данных ZINC показало, что точность реконструкции составляла ~ 70% (Jin и др. , 2018), точность восстановления составного набора данных KEGG, который мы использовали, составляла ~ 56%. Соединения KEGG содержат относительно большое количество макроциклических и длинноцепочечных соединений. Реконструкция этих соединений имеет плохую химию с JT-VAE. Это связано с тем, что оценка становится трудной, когда количество соседей в дереве соединений увеличивается или количество шагов прогнозирования увеличивается.
3.2.2 Классификация ферментативных реакций
Классы номеров EC были установлены как один и тот же класс номеров EC для каждой цифры (т.е. одна цифра: ECX; две цифры: ECX.X; три цифры: ECX.X.X). Каждый номер EC (одна цифра) имел следующее количество фрагментов данных реакции (таблица 1). Векторы признаков реакции одного и того же класса номеров EC должны быть близко распределены в пространстве признаков, потому что один и тот же тип фермента может работать для одного и того же типа структурных изменений.Чтобы исследовать представления признаков реакции, полезные для проектирования путей, комбинация древовидных и граф-латентных векторов (нормальных), древовидного латентного вектора и граф-латентного вектора JT-VAE сравнивалась на основе классификационной точности реакции. -конструкции векторов с помощью линейного дискриминантного анализа (LDA). На рисунке 7 показаны результаты классификации LDA векторов признаков реакции среди латентных векторов. Матрицы неточности из двух цифр для каждого вектора были рассчитаны путем агрегирования матриц неточностей результатов при условии, что цифра номера EC равнялась трем, а количество частей данных было больше единицы.В результате точность классификации древовидного латентного вектора была равна точности совмещенных древовидно-скрытых векторов и графов-латентных векторов. Также предполагается, что классификация вектора признаков реакции с использованием только вектора со скрытым графом была намного сложнее, чем с использованием векторов со скрытым деревом. Эти результаты показывают, что использование древовидных векторов или как древовидных, так и скрытых в графах векторов может определять характеристики каждого класса ферментов. Из приведенных выше результатов, древовидные латентные векторы были использованы для проектирования путей возможных путей.Стоимость расчетов при проектировании путей может быть снижена по сравнению с использованием латентных векторов в виде дерева и графа.
Рис. 7.
Матрицы неточностей для точности классификации каждого класса номеров EC (цифра: 2, классификатор: LDA). ( a ) Дерево и граф означает, что использовалась комбинация древовидных и граф-латентных векторов JT-VAE, ( b ) древовидный латентный вектор и ( c ) граф-латентный вектор
Рис. 7.
Матрицы неточностей для точности классификации каждого класса номеров EC (цифра: 2, классификатор: LDA).( a ) Дерево и граф означает, что использовалась комбинация древовидных и граф-латентных векторов JT-VAE, ( b ) древовидный латентный вектор и ( c ) граф-латентный вектор
Таблица 1.Количество единиц данных для каждого класса номера ЕС (одна цифра)
| Класс номера ЕС . | 1 . | 2 . | 3 . | 4 . | 5 . | 6 . | 7 . |
|---|---|---|---|---|---|---|---|
| Количество единиц данных | 3845 | 2667 | 1287 | 1158 | 488 | 344 | 4 |
| Класс номера EC . | 1 . | 2 . | 3 . | 4 . | 5 . | 6 . | 7 . |
|---|---|---|---|---|---|---|---|
| Количество единиц данных | 3845 | 2667 | 1287 | 1158 | 488 | 344 | 4 |
Количество единиц данных для каждого класса номеров EC (один цифра)
| Номер класса ЕС . | 1 . | 2 . | 3 . | 4 . | 5 . | 6 . | 7 . |
|---|---|---|---|---|---|---|---|
| Количество единиц данных | 3845 | 2667 | 1287 | 1158 | 488 | 344 | 4 |
| Класс номера EC . | 1 . | 2 . | 3 . | 4 . | 5 . | 6 . | 7 . |
|---|---|---|---|---|---|---|---|
| Количество элементов данных | 3845 | 2667 | 1287 | 1158 | 488 | 344 | 4 |
3.3 Результаты реконструкции после ферментативной реакции в латентном пространстве
Используя JT-VAE для декодирования латентных векторов продуктов, полученных путем добавления векторов признаков реакции к латентным векторам субстратов, были получены структуры продуктов, в которых произошло желаемое структурное изменение в том же классе ферментов.На рисунке 8 показан пример результатов ферментативной реакции EC1.2.1, называемой дегидрогеназой, в скрытом пространстве. «Зарегистрированная реакция (реальная)» означает, что реакция зарегистрирована в KEGG. «Виртуальный» означает, что реакция виртуально рассчитывается на компьютере. «Зарегистрированная реакция (реальная)» EC1.2.1.3, зарегистрированная в KEGG, представляет собой реакцию, в которой карбоксилат (идентификатор соединения KEGG: C00033) образуется из альдегида (идентификатор соединения KEGG: C00084). На рисунке 8 показаны три примера ферментативных реакций в латентном пространстве с использованием вектора свойств реакции EC1.2.1.3. Результаты в экс. 1 и пр. 2 на рисунке показывают, что ферментативные реакции имеют те же структурные изменения, что и «Зарегистрированная реакция (реальная)». Кроме того, результат в отл. 3 показывает, что ферментативная реакция не может происходить биологически, потому что субстраты не имеют существенных структур для катализации ферментов.
Рис. 8.
Ферментативные реакции EC1.2.1.3 в латентном пространстве
Рис. 8.
Ферментативные реакции EC1.2.1.3 в скрытом пространстве
3.4 Дизайн возможных путей
Мы разработали возможные пути при следующих условиях. Для выбора векторов признаков реакции использовали только древовидные латентные векторы. Параметры DE, то есть параметр масштабирования F и скорость кроссовера CR , были установлены равными 0,5 и 0,5 соответственно. Постоянный параметр C уравнения (5) был установлен на 1000,0. Порог ошибки Th был установлен равным 50.0. Количество популяций было установлено на 2000, а количество поколений было установлено на максимальное значение 50. Порог проверки молекулярной массы для исключения нереалистичных путей был установлен как величина изменения молекулярной массы ± 3 между основным субстратом и основной продукт зарегистрированной реакции, соответствующий выбранному номеру ЕС.
Мы подтвердили изменение количества возможных путей по отношению к размеру подмножества. На рисунке 9 показаны переходы в количестве возможных путей, когда количество повторений было установлено на 2000, а размер подмножества был изменен от 100 до 1000 с шагом 100.Каждый путь включает одну или две стадии реакции с целевым соединением. Переходы: (A) две зарегистрированные реакции, (B), (C) одна незарегистрированная реакция (два типа) и (D) зарегистрированная реакция и незарегистрированная реакция. Количество изученных возможных путей имеет тенденцию к уменьшению по мере увеличения размера подмножества. Это связано с тем, что чем больше размер подмножества, тем выше вероятность того, что подмножество будет содержать конкретную желаемую комбинацию реакций. В примерах путей, включающих одну или две стадии реакции, скорость восстановления имела тенденцию к замедлению при размерах подмножеств от 400 до 500.Число возможных путей широко варьировалось от пути к пути. Количество возможных путей имеет тенденцию к увеличению по мере увеличения количества комбинаций ферментативных реакций, отвечающих заданному порогу и имеющих сходные характеристики. Пути (A) и (B) имели меньшее количество путей-кандидатов, чем пути (C) и (D). Более того, разница между путем (C) и путем (D) была стабильной, когда размер подмножества превышал 500. Количество возможных путей уменьшалось по мере увеличения количества реакций.Следовательно, необходимо установить короткие пути для изучения многих возможных путей с помощью этого метода подмножества. Разница в количестве возможных путей связана с количеством структур, которые демонстрируют одинаковые структурные изменения.
Рис. 9.
Переход в количестве путей-кандидатов, когда количество повторов было установлено на 2000, а размер подмножества был изменен со 100 на 1000 с шагом 100. Переходы представляют собой (A) путь от C00631 к C00022, включая две зарегистрированные реакции от (B) путь от C02233 до C02845, включая одну незарегистрированную реакцию, (C) путь от C03044 до C02845, включая одну незарегистрированную реакцию и (D) путь от C00810 до C02845, включая одну зарегистрированную реакцию и незарегистрированную реакцию
Инжир.9.
Переход в количестве возможных путей, когда количество повторений было установлено на 2000, а размер подмножества был изменен со 100 на 1000 с шагом 100. Переходы представляют собой (A) путь от C00631 к C00022, включая две зарегистрированные реакции из (B ) путь от C02233 до C02845, включая одну незарегистрированную реакцию, (C) путь от C03044 до C02845, включая одну незарегистрированную реакцию и (D) путь от C00810 до C02845, включая одну зарегистрированную реакцию и незарегистрированную реакцию
3.5 Оценка возможных путей развития
3.5.1 Результаты прогноза возможности реакции
Мы применили метод прогнозирования возможности реакции с использованием ансамбля NN к ферментативным реакциям, которые включают как зарегистрированные (реальные), так и виртуальные реакции. Как описано в Разделе 3.5.1, для ввода использовались вектор признаков реакции и вектор субстрата, скрытый в виде дерева и графа. Мы рассмотрели следующие два условия относительно входных данных.
Независимо от того, является ли пара ферментных реакций, составляющая вектор признаков реакции, реальной или виртуальной.
Является ли субстрат вектором признаков реакции.
Таким образом, мы сначала подготовили следующие четыре типа наборов данных для обучения нейронных сетей.
«Реальный» набор данных, состоящий из реального вектора признаков ферментативной реакции, зарегистрированного в KEGG, и латентного вектора субстрата, используемого для расчета вектора признаков реакции ферментативной реакции (Реальная пара, Субстрат в).
Набор данных «Virtual-1», состоящий из реального вектора признаков ферментативной реакции, полученного из KEGG, и латентных векторов субстратов, не используемых для расчета вектора признаков реакции ферментативной реакции (Реальная пара, Субстрат вне).
Набор данных Virtual-2, состоящий из виртуального вектора признаков ферментативной реакции, состоящего из латентного вектора субстрата и продукта, которые были выбраны случайным образом, и латентного вектора субстрата, используемого для вычисления вектора признаков реакции ферментативные реакции (Виртуальная пара, Субстрат в).
Набор данных «Virtual-3», состоящий из виртуального вектора характеристик ферментативной реакции, состоящего из латентного вектора субстрата и продукта, которые были выбраны случайным образом, и латентного вектора субстрата, который не использовался для расчета реакции -вектор функций (виртуальная пара, выход подложки).
Набор данных Real содержал 9794 единицы данных ферментативных реакций, зарегистрированных в KEGG. Кроме того, количество наборов данных каждого виртуального типа было 10.Каждый виртуальный набор данных содержит 10 000 единиц данных. Таким образом, один обучающий набор данных, объединяющий реальные и виртуальные данные, состоял из 19 794 единиц данных. В ходе обучения было получено 150 весов при выполнении 5-кратной перекрестной проверки для каждого набора данных.
Каждая сеть имеет три полносвязных средних уровня (64, 32, 8). Все функции активации были установлены как выпрямленные линейные единицы. Каждая модель выводит 0 или 1 для каждого входа. Было взято среднее из этих 150 выходов, и, наконец, значение возможности реакции было рассчитано от 0.От 0 до 1.0.
В таблице 2 перечислены результаты среднего и стандартного отклонения прогноза возможности реакции для каждого типа данных. Мы подтвердили, что оценки реальных и виртуальных данных существенно различались. Среднее значение «реальных» данных было близко к 1,0. Однако значения «Virtual-1» и «Virtual-2» были близки или меньше 0,5. Значения данных «Virtual-3» были очень маленькими. Чем больше реалистичных элементов было включено, тем выше была вероятность реакций, а виртуальные данные не были полностью нулевыми.0. Это разумный результат, потому что реакция, которая может быть определена как реальная, фактически включается при оценке незарегистрированной реакции.
Таблица 2.Результаты прогноза возможности реакции (макс: 1,0; мин: 0,0)
| . | Подложка в . | Субстрат вне . |
|---|---|---|
| Реальная пара | 0,99 ± 0,02 | 0,54 ± 0,23 |
| Виртуальная пара | 0.35 ± 0,17 | 0,09 ± 0,12 |
| . | Подложка в . | Субстрат вне . |
|---|---|---|
| Реальная пара | 0,99 ± 0,02 | 0,54 ± 0,23 |
| Виртуальная пара | 0,35 ± 0,17 | 0,09 ± 0,12 |
Результаты прогнозирования возможности реакции (макс. : 1,0; мин: 0,0)
| . | Подложка в . | Субстрат вне . |
|---|---|---|
| Реальная пара | 0,99 ± 0,02 | 0,54 ± 0,23 |
| Виртуальная пара | 0,35 ± 0,17 | 0,09 ± 0,12 |
| . | Подложка в . | Субстрат вне . |
|---|---|---|
| Реальная пара | 0.99 ± 0,02 | 0,54 ± 0,23 |
| Виртуальная пара | 0,35 ± 0,17 | 0,09 ± 0,12 |
3.5.2 Результаты оценки пути кандидата
Прогнозирование возможности реакции было подтверждено с использованием части пути «гликолиза». В частности, значение осуществимости каждого пути-кандидата, полученного с использованием метода прогнозирования возможности реакции, было проверено, когда пути были разработаны на основе условия, что все векторы признаков реакции используются в пути от α -d-глюкозо-6-фосфат ( KEGG соединение ID: C00668) в глицеральдегид-3-фосфат (KEGG соединение ID: C00118).То есть выбранными ферментативными реакциями были EC5.3.1.9, EC2.7.1.1 и EC4.1.2.13. На рис. 10 показаны результаты значений возможных путей для возможных путей. Следует отметить, что после реконструкции соединений некоторые пути могут быть удалены путем сокращения пути на основе изменения молекулярных масс. Мы подтвердили, что зарегистрированный путь имеет наивысшее значение выполнимости и что оценка пути указывает на то, что сокращение пути с использованием значения вероятности реакции также может быть применено.
Рис. 10.
Результаты оценки пути-кандидата. Использовали путь от α -d-глюкозо-6-фосфата (KEGG соединение ID: C00668) до глицеральдегид-3-фосфата (KEGG соединение ID: C00118). Выбранными ферментативными реакциями были EC5.3.1.9, EC2.7.1.1 и EC4.1.2.13. Всего было шесть комбинаций. Каждая линия, обозначающая каждый вектор-признак реакции, имеет разный цвет, а толщина линии соответствует значению возможности каждой реакции
Рис.10.
Результаты оценки пути кандидата. Использовали путь от α -d-глюкозо-6-фосфата (KEGG соединение ID: C00668) до глицеральдегид-3-фосфата (KEGG соединение ID: C00118). Выбранными ферментативными реакциями были EC5.3.1.9, EC2.7.1.1 и EC4.1.2.13. Всего было шесть комбинаций. Каждая линия, обозначающая вектор каждого признака реакции, имеет разный цвет, а толщина линии соответствует значению возможности каждой реакции
3,6 Исследование возможных путей
Мы применили предложенный метод к незарегистрированным путям, чтобы проверить его работоспособность.На рисунке 11 показаны две ферментативные реакции путей валидации, которые являются незарегистрированными путями производства целевого соединения 2-бутанона (KEGG Compound ID: C02845), о которых сообщалось в предыдущих исследованиях (Chen et al. , 2015; Srirangan et al. ). , 2016).
Рис. 11.
Результаты изучения возможных путей. Путь от пирувата (идентификатор соединения KEGG: C00022) к 2-бутанону (идентификатор соединения KEGG: C02845) и путь от ацетил-КоА (идентификатор соединения KEGG: C00024) до 2-бутанона описаны в Srirangan et al. (2016) и Chen et al. (2015) соответственно, но обе реакции прекурсоров на 2-бутанон в KEGG не зарегистрированы. Незарегистрированные реакции представлены красными пунктирными линиями. Более того, v r s являются значениями вероятности реакции. Обе реакции были исследованы с использованием предложенной методики.
Рис. 11.
Результаты изучения возможных путей. Путь от пирувата (идентификатор соединения KEGG: C00022) к 2-бутанону (идентификатор соединения KEGG: C02845) и путь от ацетил-КоА (идентификатор соединения KEGG: C00024) до 2-бутанона описаны в Srirangan et al. (2016) и Chen et al. (2015) соответственно, но обе реакции прекурсоров на 2-бутанон в KEGG не зарегистрированы. Незарегистрированные реакции представлены красными пунктирными линиями. Более того, v r s являются значениями вероятности реакции. Обе реакции были исследованы с использованием предложенной методики.
Сначала мы исследовали векторы признаков реакции, наиболее близкие к двум типам зарегистрированных незарегистрированных реакций в каждом трехзначном классе ферментативных реакций.Затем мы проверили, можно ли исследовать пути с помощью предлагаемой техники. Каждый вектор признаков реакции вычисляли по разнице между латентным вектором каждого целевого соединения и векторами предшественника. Конструирование путей кандидатов проводилось с использованием только древовидного вектора JT-VAE. Оценка пути и каждое значение возможности реакции ферментативных реакций также выводились с помощью метода прогнозирования возможности реакции.
Мы исследовали возможные пути, соединяющие метаболические пути, как показано на рисунке 11.Путь валидации A был описан Srirangan et al. (2016). Этот путь включает незарегистрированную ферментативную реакцию, в которой 2-бутанон образуется из 3-оксопентаноата (идентификатор соединения KEGG: C02233). Номер ЕС соответствующей реакции 4.1.1.4. Ферментативная реакция EC4.1.1.4, зарегистрированная в KEGG, представляет собой реакцию, из которой ацетон (идентификатор соединения KEGG: C00207) образуется из ацетоацетата (идентификатор соединения KEGG: C00164). Этот путь сообщает о пути, включающем соединение с КоА, но поскольку сложно нацелить длинное соединение, такое как КоА, с JT-VAE, мы исследовали его на основе предшественника, 3-оксопентаноата.А именно, мы применили предложенный метод к пути, когда 2-бутанон был установлен в качестве целевого соединения, а 3-оксопентаноат в качестве предшественника был установлен в качестве исходного соединения. Путь валидации B был описан Chen et al. (2015). Этот путь включает незарегистрированную ферментативную реакцию, в результате которой 2-бутанон образуется из 2,3-бутандиола (идентификатор соединения KEGG: C003044). Номер ЕС соответствующей реакции 4.2.1.28. Ферментативная реакция EC4.2.1.28, зарегистрированная в KEGG, представляет собой реакцию, в результате которой пропаналь (идентификатор соединения KEGG: C00479) образуется из пропан-1,2-диола (идентификатор соединения KEGG: C00583).Для подтверждения пути B исследование пути было проведено с помощью предложенной методики с использованием ацетоина (KEGG Compound ID: C00810), который является предшественником предшественника, в качестве исходного соединения. Мы подтвердили, что каждый вектор признаков реакции, генерируемый субстратом и продуктом, описанными в каждом исследовании, был очень похож на вектор признаков реакции числа ЕС, описанный в этих исследованиях. В обеих ферментативных реакциях номер ЕС наиболее сходного вектора-признака реакции в соответствующем классе номеров ЕС (три цифры) совпадал с номером, описанным в этих статьях.Более того, с помощью предлагаемой техники можно изучить возможные пути, включая потенциальные пути, о которых сообщалось в предыдущем исследовании (Chen et al. , 2015; Srirangan et al. , 2016), как показано на Рисунке 11, т. Е. Красным пунктирные линии. Путь B от пирувата до 2-бутанона включает четыре или пять реакций. Однако, когда 2-бутанон был исследован в качестве целевого соединения, правильный путь был получен для одно- или двухстадийных реакций на 2-бутанон. А именно, мы получили правильные пути, когда в качестве исходного соединения были выбраны 2,3-бутандиол или ацетоин.Мы подтвердили, что если количество реакций равно трем или более, вероятность того, что правильные реакции были включены в подмножество, уменьшается; таким образом, исследование становится трудным.
4 Обсуждение и выводы
Мы предложили метод исследования возможных путей, который включает (i) представление реакции с использованием скрытого химического пространства для ферментативной реакции в компьютерной системе, (ii) проектирование пути-кандидата с использованием алгоритма DE путем объединения потенциальных ферментативных реакций и (iii) ) оценка пути с использованием основанного на NN метода прогнозирования возможности реакции для определения значений осуществимости пути возможных путей.Мы применили предложенный метод к незарегистрированным путям, связанным с производством 2-бутанона. Предлагаемый метод исследовал возможные пути, включая незарегистрированные ферментативные реакции.
Из результатов, показанных на фиг. 8 и 11, можно сказать, что такое же структурное изменение, как и соответствующая ферментативная реакция, может происходить путем добавления вектора-признака реакции к латентному вектору субстрата. Как показано на Рисунке 8 для «Virtual 3», отклонение реакций от правил ферментативных реакций было подтверждено, поскольку правила ферментативных реакций не применялись, хотя степень свободы представления реакции была высокой.Мы удалили пути, включая такие реакции, на основе изменения молекулярной массы. С гибридным методом, применяющим минимальные правила ферментативной реакции для представления реакции, можно ожидать более точного решения этой проблемы.
Древовидный латентный вектор JT-VAE, используемый для исследования путей, был полезен для классификации ферментных реакций и дизайна путей, подтверждая, что он может улавливать субстратную специфичность ферментных реакций. Это связано с тем, что метод дерева характеристик (Rarey and Dixon, 1998), который рассматривает подструктуры как фрагменты, может фиксировать сходство изменений в общей структуре магистрали.Более того, при проектировании возможных путей бинарный алгоритм DE просто применялся к проблеме НЛП, размер которой был большим в сочетании с методом подмножества. Это очень эффективный метод исследования путей, включающих одну или две реакции. А именно, возможные пути могут быть исследованы, когда предшественник целевого соединения или соединение до предшественника было установлено в качестве исходного соединения. Использование метода подмножества вызвало проблему, заключающуюся в том, что эффективное решение не могло быть предоставлено, если соответствующая реакция не была включена из-за увеличения количества реакций.Для решения этой проблемы эффективны предварительная кластеризация признаков реакции и применение многоэтапного поиска с использованием центрального вектора. Это позволяет выполнять поиск, нацеленный на все реакции, сохраняя при этом эффективность поиска. Также эффективен метод, с помощью которого БД векторов признаков реакции формируется в древовидную структуру.
Судя по результатам на Рисунке 11, значения осуществимости возможных путей с использованием метода прогнозирования возможности реакции на основе NN были близки к 1,0 для фактических путей и реакций, не зарегистрированных в KEGG, но описанных в документе.Значения были ниже для незарегистрированных реакций, о которых не сообщалось. Таким образом, нам удалось создать метод прогнозирования возможности реакции на основе БД зарегистрированных реакций. При оценке пути необходимо более точно оценивать ферментативные реакции, собирая данные о ферментативных реакциях в других базах данных и статьях. Эффективно не только судить о наличии зарегистрированной ферментативной реакции, но также проводить тренировку с таким показателем, как физическая величина, относящаяся к ферментативной реакции. Например, можно включить такие индикаторы, как токсичность и естественность.
Что касается будущих проблем, связанных с химическими VAE, необходим метод повышения точности составной реконструкции и работы с соединениями, исключенными в этой статье. Существует потребность в технологии, которая может использовать длинноцепочечные соединения, которые имеют длинные строки символов SMILES, соединения, содержащие макроциклы, и соединения, представленные ионными связями, которые нельзя игнорировать в метаболических путях. В качестве современной техники была предложена гиперграф-грамматика для химических структур (Kajino, 2019).Этот метод имеет более высокую точность составной реконструкции, чем JT-VAE. Мы усовершенствуем предложенную технику, чтобы можно было более точно исследовать многоступенчатые пути.
Благодарности
Мы хотели бы поблагодарить профессора Мичихиро Араки за его ценные комментарии.
Финансовая поддержка : Это исследование основано на результатах, полученных в рамках проекта, заказанного Организацией по развитию новой энергетики и промышленных технологий (NEDO).
Конфликт интересов : не объявлен.
Доступность данных
Наборы данных, использованные в этом исследовании, можно получить у соответствующего автора, Т. Фуджи ([email protected]), по разумному запросу.
Список литературы
Араки
М.
и другие. (2014
)M-path: компас для навигации по потенциальным метаболическим путям
.Биоинформатика
,31
,905
—911
.Баттити
р.
,Colla
A.M.
(1994
)Демократия в нейронных сетях: схемы голосования для классификации
.Нейронные сети
,7
,691
—707
.Caspi
р.
и другие. (2018
)База данных метаболических путей и ферментов MetaCyc
.Nucleic Acids Res
.,46
,D633
—D639
.Чен
З.
и другие. (2015
)Метаболическая инженерия клебсиелл пневмонии для производства 2-бутанона из глюкозы
.PLoS One
,10
,e0140508
.Чой
К.Р.
и другие. (2019
)Системные стратегии метаболической инженерии: интеграция систем и синтетической биологии с метаболической инженерией
.Trends Biotechnol
.,37
,817
—837
.Дегтяренко
К.
и другие. (2007
)Chebi: база данных и онтология химических объектов, представляющих биологический интерес
.Nucleic Acids Res
.,36
(Suppl. 1
),D344
—D350
.Делепин
B.
и другие. (2018
)Retropath3. 0: рабочий процесс ретросинтеза для инженеров-метаболиков
.Metabolic Eng
.,45
,158
—170
.Гомес-Бомбарелли
р.
и другие. (2018
)Автоматический химический дизайн с использованием непрерывного представления молекул на основе данных
.ACS Cent. Sci
.,4
,268
—276
.Хадади
N.
,Hatzimanikatis
V.
(2015
)Разработка компьютерных инструментов ретробиосинтеза для разработки синтетических путей de novo
.Curr. Opin. Chem. Биол
.,28
,99
—104
.Jin
W.
и другие. (2018
) Вариационный автокодер дерева соединений для построения молекулярных графов. В: Международная конференция по машинному обучению , Stockholmsmässan, Стокгольм, Швеция, 10 июля 2018 г. — 15 июля 2018 г., стр.2328
—2337
.Каджино
H.
(2019
) Грамматика молекулярного гиперграфа в применении к молекулярной оптимизации.В: Международная конференция по машинному обучению , Лонг-Бич, Калифорния, США, 10 июня 2019 г. — 15 июня 2019 г., стр.3183
—3191
.Канехиса
М.
,Goto
S.
(2000
)KEGG: Киотская энциклопедия генов и геномов
.Nucleic Acids Res
.,28
,27
—30
.Ким
С.
и другие. (2016
)База данных веществ и соединений PubChem
.Nucleic Acids Res
.,44
,D1202
—D1213
.Кумар
A.
и другие. (2018
)Дизайн пути с использованием шагов de novo через неизведанные биохимические пространства
.Nat. Коммуна
.,9
,184
.Куснер
M.J.
и другие. (2017
) Грамматический вариационный автоэнкодер. В: Международная конференция по машинному обучению , Сидней, Австралия, 6 августа 2017 г. — 11 августа 2017 г., стр.1945
—1954
.Моретти
S.
и другие. (2016
)Metanetx / mnxref – согласование метаболитов и биохимических реакций для объединения метаболических сетей в масштабе генома
.Nucleic Acids Res
.,44
,D523
—D526
.Мория
Ю.
и другие. (2010
)Pathpred: сервер прогнозирования метаболических путей, катализируемых ферментами.
.Nucleic Acids Res
.,38
(Suppl.2
),W138
—W143
.Рэрей
М.
,Диксон
J.S.
(1998
)Деревья признаков: новая мера молекулярного сходства, основанная на сопоставлении деревьев
.J. Comput. Помощь Мол. Des
.,12
,471
—490
.Шриранган
К.
и другие. (2016
)Engineering Escherichia coli для микробиологического производства бутанона
.заявл. Environ. Microbiol
.,82
,2574
—2584
.стерлингов
т.
,Irwin
J.J.
(2015
)Цинк 15 — открытие для каждого
.J. Chem. Инф. Модель
.,55
,2324
—2337
.Сторн
р.
,Цена
к.
(1997
)Дифференциальная эволюция — простая и эффективная эвристика для глобальной оптимизации в непрерывных пространствах
.J. Global Optim
.,11
,341
—359
.Ван
Л.
и другие. (2017
)Обзор вычислительных инструментов для проектирования и реконструкции метаболических путей
.Synth. Syst. Biotechnol
.,2
,243
—252
.© Автор (ы) 2020. Опубликовано Oxford University Press.
Это статья в открытом доступе, распространяемая в соответствии с условиями лицензии Creative Commons Attribution License (http: // creativecommons.org / licenses / by / 4.0 /), который разрешает неограниченное повторное использование, распространение и воспроизведение на любом носителе при условии правильного цитирования оригинальной работы.Произошла ошибка при установке пользовательского файла cookie
Этот сайт использует файлы cookie для повышения производительности. Если ваш браузер не принимает файлы cookie, вы не можете просматривать этот сайт.
Настройка вашего браузера для приема файлов cookie
Существует множество причин, по которым cookie не может быть установлен правильно.Ниже приведены наиболее частые причины:
- В вашем браузере отключены файлы cookie. Вам необходимо сбросить настройки своего браузера, чтобы он принимал файлы cookie, или чтобы спросить вас, хотите ли вы принимать файлы cookie.
- Ваш браузер спрашивает вас, хотите ли вы принимать файлы cookie, и вы отказались. Чтобы принять файлы cookie с этого сайта, нажмите кнопку «Назад» и примите файлы cookie.
- Ваш браузер не поддерживает файлы cookie. Если вы подозреваете это, попробуйте другой браузер.
- Дата на вашем компьютере в прошлом.Если часы вашего компьютера показывают дату до 1 января 1970 г., браузер автоматически забудет файл cookie. Чтобы исправить это, установите правильное время и дату на своем компьютере.
- Вы установили приложение, которое отслеживает или блокирует установку файлов cookie. Вы должны отключить приложение при входе в систему или проконсультироваться с системным администратором.
Почему этому сайту требуются файлы cookie?
Этот сайт использует файлы cookie для повышения производительности, запоминая, что вы вошли в систему, когда переходите со страницы на страницу.Чтобы предоставить доступ без файлов cookie потребует, чтобы сайт создавал новый сеанс для каждой посещаемой страницы, что замедляет работу системы до неприемлемого уровня.
Что сохраняется в файле cookie?
Этот сайт не хранит ничего, кроме автоматически сгенерированного идентификатора сеанса в cookie; никакая другая информация не фиксируется.
Как правило, в файле cookie может храниться только информация, которую вы предоставляете, или выбор, который вы делаете при посещении веб-сайта.Например, сайт не может определить ваше имя электронной почты, пока вы не введете его. Разрешение веб-сайту создавать файлы cookie не дает этому или любому другому сайту доступа к остальной части вашего компьютера, и только сайт, который создал файл cookie, может его прочитать.
Обнаружен скрытый метаболический путь
Прежде чем читать отчет исследовательской группы Сиднея Кусту, опубликованный в недавнем выпуске PNAS (1), я серьезно сомневался, что открытие нового пути катаболизма пиримидина у Escherichia coli K12 было чем-то это было возможно и сегодня.После всех лет исчерпывающих генетических и биохимических исследований, почти десятилетия после завершения генома (2), после бесчисленных усилий по аннотации и повторной аннотации, метаболического моделирования (3) и систематических исследований функциональной геномики (4-6), полностью новый путь был найден? Хотя функциональные роли многих генов E. coli (по крайней мере 20%) все еще неизвестны, эти оставшиеся гены – в значительной степени воспринимаются как потенциальные второстепенные игроки в некоторых малоизвестных областях метаболизма или, что еще более вероятно, в неметаболических процессах. процессы.Следовательно, цепочка из восьми генов y , кодирующих весь путь в самом центре метаболической карты E. coli , казалась слишком большим скелетом, чтобы спрятать его в шкафу.
Как могли не только конкретные гены, но и само существование этого пути ускользнуть от более ранних исследований? Одно из возможных объяснений состоит в том, что «пиримидиновые кольца могут использоваться в качестве единственного источника азота при комнатной температуре, но не при 37 ° C» (1). По-прежнему остается вопрос: почему среди множества успешных поисков отсутствующих генов (7) все мощные методы биоинформатики не смогли обнаружить этот недостающий путь? Вероятное объяснение (в одной из его многочисленных версий) состоит в том, что «чтобы найти черную кошку в темной комнате, мы должны хотя бы знать, что она там прячется.Действительно, ранние физиологические данные (8), если они не были зафиксированы в публичных архивах, таких как Киотская энциклопедия генов и геномов (KEGG) (www.genome.ad.jp/kegg) или EcoCyc, Энциклопедия генов Escherichia coli K-12 Genes and Metabolism (http://ecocyc.org) оставалась недоступной для аннотаторов генома, что мешало им задавать правильные вопросы. С другой стороны, специалисты в данной области часто не знают об инструментах биоинформатики, которые помогли бы им решить этот вопрос.Рассказ о скрытом пути катаболизма пиримидина (1) иллюстрирует силу объединения обоих миров. Я вернусь к этой теме после краткого обсуждения конкретных значений этого исследования.
Сложный механизм катаболизма пиримидина, обнаруженный Кусту и соавторами (1), является важным дополнением к сети утилизации азота в E. coli . Этот механизм, классический оперон (теперь называемый опероном rut , см. Рис. 1). A ), находится под регуляцией нового репрессора RutR (бывший ген ycdC ), конкурирующего с NtrC, роль которого в качестве глобального «диспетчера азота» была выяснена группой Кусту ранее (9).Основная физиологическая роль этого пути не очевидна, по крайней мере, для членов этой группы, которые подозревают, что он может вносить больший вклад в глобальную клеточную регуляцию, чем в простое питание (1). Тем не менее, в лучших катаболических традициях оперон rut снабжен транспортером RutG (бывший ген ycdG ). Конечный продукт этого пути, гидроксипропионат (или 2-метил-гидроксипропионат), является еще одной новинкой, отличающей его от обоих «устаревших» путей, описанных у других видов и отраженных на карте KEGG (www.genome.jp/dbget-bin/www_bget?path:rn00240). Отличие от восстановительного пути также подтверждается сравнением генов, в то время как генетические данные для окислительного пути, описанного в ранних исследованиях коринебактерий и других видов, отсутствуют (10–12).
Рисунок 1.Кластеризация на хромосоме генов, участвующих в новом пути (гены A , rut ) и в ранее охарактеризованном восстановительном пути ( B , гены pyd ) катаболизма пиримидина.Названия генов приведены в таблице 1. Соответствующие цвета (на каждой панели) соответствуют ортологам генов. Уникальной особенностью оперона pyd в P. aeruginosa (но не у других Pseudomonadales) является наличие генов, кодирующих цитозинпермеазу ( codB , гомолог PydP ) и цитозиндезаминазу ( codA ). , который будет направлять цитозин по тому же пути утилизации.
Таблица 1.Встречаемость генов, участвующих в катаболизме пиримидинов, в полностью секвенированных геномах
Сравнительный анализ генома позволил Кусту и его коллегам (1) расширить влияние своих выводов за пределы модельной системы E. coli , как показано в таблице 1, сжатой форме подсистемы утилизации пиримидина, скомпилированной с семенной платформой (http : //theseed.uchicago.edu/FIG/index.cgi). Подсистемный подход к аннотации генома и анализу путей позволяет нам фиксировать существующие знания о биологических процессах и надежно проецировать их на всю коллекцию различных видов с полностью секвенированными геномами (13).Помимо установления того, какие организмы реализуют тот или иной функциональный вариант подсистемы (например, восстановительный путь или путь rut ), этот подход помогает выявить пробелы в знаниях (отсутствующие гены) и потенциальных новых игроков (предсказанные гены).
Гены пути rut сгруппированы на хромосоме у всех видов (см. Таблицу 1 и Рис. A ), что является убедительным доказательством их функциональной связи (14). Регулятор RutR и большинство ферментативных компонентов являются консервативными, за исключением предполагаемой редуктазы RutE, возможного объекта смещения неортологичного гена у нескольких видов помимо Enterobacteria.Отсутствие ортологов RutG у некоторых видов может отражать альтернативную транспортную систему или исключительную роль пути rut в контроле внутреннего пула пиримидинов.
Ядро восстановительного пути состоит из трех ферментативных стадий: ( i ) дигидропиримидиндегидрогеназа, ( ii ) дигидропиримидиназа и ( iii ) β-уреидопропионаза, исторически охарактеризованная в контексте метаболических нарушений у человека ( 15). Бактериальные гены, кодирующие функциональные гомологи этих ферментов [ pydA , pydB и pydC , охарактеризованные в Brevibacillus agri (16)], образуют хромосомный кластер, консервативный у различных видов (см. Рис.1 В ). Наиболее поразительным наблюдением при сравнении этих двух путей является то, что единственный ген, общий для них, — это регулятор rutR (17), который является консервативным и тесно связан с обоими оперонами. Хотя это сохранение может быть объяснено совместным использованием эффекторов (пиримидинов), эволюционный сценарий, объясняющий это наблюдение, не очевиден. Единственная копия rutR в Agrobacterium tumefaciens , несущая оба оперона, сгруппирована с опероном pyd (см.рис.1), что также является единственным примером неполного восстановительного пути. Хотя отсутствие pydA в этом организме можно объяснить неортологическим смещением, уникальное сосуществование двух путей может вызвать другие объяснения.
Транспортные механизмы различаются между путями и между группами видов. На основании кластеризации хромосом можно выделить два типа транспортных систем для редуктивного пути (см. Рис. 1). В ). Пермеаза PydP, характерная для β- и γ-протеобактерий (например,g., PA0443 в Pseudomonas aeruginosa ), по-видимому, функционально заменяется кассетой ABC у α-протеобактерий. Дополнительный компонент восстановительного пути, называемый здесь PydX (например, PA0440 в P. aeruginosa ), может быть выведен на основании данных о кластеризации хромосом (14) и слиянии белков (18). Последнее обеспечивается наблюдаемой гомологией между PydX и N-концевым доменом дигидропиримидиндегидрогеназы человека, который, как известно, обеспечивает цепь переноса электрона для каталитического С-концевого домена (19), гомолога бактериального PydA.
Несмотря на важность конкретных результатов, обсужденных выше, влияние опубликованного исследования (1) выходит за рамки этой интересной подсистемы, предоставляя нам замечательную иллюстрацию силы комбинирования методов функциональной геномики, таких как анализ микроматриц и фенотипический скрининг, со сравнительными геномика для картирования совершенно новых путей. Анализируя путь rut ретроспективно, можно заметить ряд важных ключей, которые дает только биоинформатика: ( i ) кластеризация хромосом и совместное появление генов rut указывает на то, что они могут принадлежать к одному и тому же пути, ( ii ) основанная на гомологии аннотация RutG как переносчика азотистых оснований указывает на возможную роль этого пути в использовании пиримидинов или пуринов, и ( iii ) совместное использование ортологов RutR между оперонами rut и pyd указывает на возможное участие с катаболизмом пиримидина.Эти подсказки в сочетании с скудными доказательствами в литературе могут вызвать гипотезу, подлежащую целенаправленной экспериментальной проверке. Этот анализ (независимо от того, насколько он спекулятивен по отношению к уже завершенному исследованию) направлен на то, чтобы подчеркнуть огромные возможности, предоставляемые сравнительным анализом нескольких секвенированных геномов. Растущее число успешных примеров (среди которых рассказ о скрытом пути rut является одним из самых ярких) и растущее понимание инструментов сравнительной геномики побудят многие исследовательские группы изучить эти возможности, тем самым ускоряя темпы развития генов и геномов. открытие пути.
Сноски
- Электронная почта: osterman burnham.org
См. Сопутствующую статью на странице 5114 в выпуске 13 тома 103.
Автор (ы): A.O. написал газету.
Заявление о конфликте интересов: о конфликте интересов не сообщалось.
- © 2006 Национальная академия наук США
Путь метаболизма гликолиза
Гликолиз — это термин, используемый для описания метаболического пути, включающего расщепление глюкозы на пируват и энергию, используемую для образования аденозинтрифосфата (АТФ) и восстановленного никотинамидадениндинуклеотида (НАДН).
Путь происходит почти у всех организмов и не зависит от кислорода, хотя продукты гликолиза иногда разлагаются с помощью атмосферного кислорода.
Следующее уравнение суммирует ряд реакций, которые происходят в пути гликолиза:
C 6 H 12 O 6 + 2 NAD + + 2 ADP + 2 P -> 2 пировиноградная кислота, (CH 3 (C = O) COOH) + 2 ATP + 2 NADH + 2 часа +
Существует ряд этапов, которые необходимо выполнить для того, чтобы произошел гликолиз, которые можно разделить на две основные части: подготовительная фаза и фаза погашения.
Инфографика аэробного дыхания и процесса гликолиза. Кредит изображения: VectorMine / Shutterstock.com
Подготовительный этап
Также известная как инвестиционная фаза, подготовительная фаза включает потребление АТФ для запуска метаболического процесса. В эту фазу входят следующие шаги:
1. Синтез фосфатного эфира: Эндотермическая реакция, катализируемая гексокиназой, использует энергию АТФ для синтеза фосфатного эфира в молекулу глюкозы в положении C-6.
2. Изомеризация: Реакция, катализируемая фосфоглюкоизомеразой, превращает глюкозо-6-фосфат в изомер фруктозо-6-фосфат.
3. Синтез фосфатного эфира: Эндотермическая реакция, катализируемая фосфофруктокиназой, использует энергию АТФ для синтеза фосфатного эфира с молекулой фруктозо-6-фосфата в положении C-1.
4. Расщепленная молекула: Молекула фруктозо-1,6-дифосфата подвергается реакции обратной альдольной конденсации, катализируемой альдолазой, с образованием двух углеродных соединений, альдегида и кетона.
Из двух молекул дигидроксиацетонфосфата, образовавшихся в результате этого расщепления, одна сразу переходит в фазу погашения, а другая претерпевает другую реакцию и затем переходит во вторую фазу. Последний шаг:
5. Изомеризация: Реакция, катализируемая триозофосфатизомеразой, превращает дигидроксиацетонфосфат в глицеральдегид-3-фосфат.
Этап окупаемости
По завершении реакций на подготовительной фазе фаза погашения может начинаться и проходить до завершения дважды, сначала с молекулой глицеральдегида, а затем с молекулой глицеральдегид-3-фосфата.Реакции следующие:
1. Синтез фосфатного эфира: реакция окисления альдегида, катализируемая глицеральдегид-3-фосфатом, приводит к образованию дифосфоглицерата.
2. Гидролиз фосфата: реакция гидролиза, катализируемая фосфоглицеринкиназой, образует кислоту, ион фосфата и АТФ.
3. Изомеризация: движение фосфатной группы, катализируемое фосфоглицератмутазой, происходит из положения 3 в положение 2.
4. Дегидратация спирта: реакция дегидратации, катализируемая енолазой, удаляет спирт (-OH) и (-H) из атомов углерода C-3 и C-2.
5. Гидролиз фосфатного эфира: конечная реакция гидролиза, катализируемая пировиноградной киназой, стабилизирует молекулу и образует кислоту, фосфат-ион и АТФ.
По завершении стадии погашения путь гликолиза завершен.
Этапы гликолиза | Клеточное дыхание | Биология | Khan Academy Играть
Список литературы
Дополнительная литература
6.1С: Метаболические пути — Биология LibreTexts
- Последнее обновление
- Сохранить как PDF
- Ключевые моменты
- Ключевые термины
- Метаболические пути
- Анаболические пути
- Катаболические пути
- Важность ферментов
Анаболический путь требует энергии и строит молекулы, в то время как катаболический путь производит энергию и расщепляет молекулы.
Цели обучения
- Описать два основных типа метаболических путей
Ключевые моменты
- Метаболический путь — это серия химических реакций в клетке, которые создают и разрушают молекулы для клеточных процессов.
- Анаболические пути синтезируют молекулы и требуют энергии.
- Катаболические пути расщепляют молекулы и производят энергию.
- Поскольку почти все метаболические реакции происходят не спонтанно, белки, называемые ферментами, помогают облегчить эти химические реакции.
Ключевые термины
- катаболизм : деструктивный метаболизм, обычно включающий выделение энергии и расщепление материалов
- фермент : глобулярный белок, катализирующий биологическую химическую реакцию
- анаболизм : конструктивный метаболизм тела в отличие от катаболизма
Метаболические пути
Процессы производства и расщепления углеводных молекул иллюстрируют два типа метаболических путей.Метаболический путь — это последовательный ряд взаимосвязанных биохимических реакций, которые преобразуют молекулу или молекулы субстрата через ряд промежуточных продуктов метаболизма, в конечном итоге приводя к конечному продукту или продуктам. Например, один путь метаболизма углеводов расщепляет большие молекулы на глюкозу. Другой метаболический путь может превращать глюкозу в большие молекулы углеводов для хранения. Первый из этих процессов требует энергии и называется анаболическим. Второй процесс производит энергию и называется катаболическим.Следовательно, метаболизм состоит из этих двух противоположных путей:
- Анаболизм (построение молекул)
- Катаболизм (разрушение молекул)
Анаболические пути
Анаболические пути требуют ввода энергии для синтеза сложных молекул из более простых. Одним из примеров анаболического пути является синтез сахара из CO 2 . Другие примеры включают синтез крупных белков из строительных блоков аминокислот и синтез новых цепей ДНК из строительных блоков нуклеиновых кислот. Эти процессы имеют решающее значение для жизни клетки, происходят постоянно и требуют энергии, обеспечиваемой АТФ и другими высокоэнергетическими молекулами, такими как НАДН (никотинамидадениндинуклеотид) и НАДФН.
Катаболические пути
Катаболические пути включают разложение сложных молекул на более простые, высвобождая химическую энергию, хранящуюся в связях этих молекул. Некоторые катаболические пути могут захватывать эту энергию для производства АТФ, молекулы, используемой для питания всех клеточных процессов. Другие запасающие энергию молекулы, такие как липиды, также расщепляются посредством аналогичных катаболических реакций, высвобождая энергию и производя АТФ.